Total synthesis is sometimes the only way to explore the chemical space around a natural product
It often feels that chemists’ talk of biological activity at the start of total synthesis papers is just boilerplate. But natural products continue to be an important source of new drugs. In therapeutic areas such as antibiotics and oncology, natural products and their derivatives make up more than half of approved drugs.1 Big Pharma’s enthusiasm for natural product chemistry has waxed and waned over the years, but with the spectre of a post-antibiotic era looming, chemists are again looking to nature for inspiration in the fight against drug-resistant bacteria.
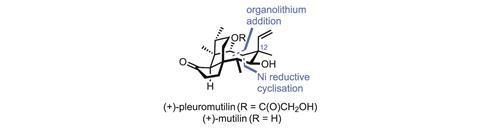
The diterpene fungal metabolite (+)-pleuromutilin has already successfully served as a starting point for antibiotic development. Starting from natural pleuromutilin, GlaxoSmithKline (GSK) created retapamulin, a powerful antibiotic that was approved by the US Food and Drug Administration (FDA) in 2007. Lefamulin, another pleuromutilin-derived compound from Nabriva Therapeutics, is also in clinical trials. However, while such semi-synthetic approaches save time and money by shifting work from chemists to fermenters, they do have downsides. One major limitation is that teams are often stuck with the majority of the natural product structure, as it’s just too difficult to make big changes. Indeed, both the GSK and Nabriva teams opted to simply swap out the molecule’s peripheral acyl side chain, leaving a vast amount of chemical space – and many potential new drugs – unexplored. This is unfortunate, as tweaks to the molecule’s eight-membered ring, such as inverting the C-12 stereocentre, can profoundly alter its bioactivity. However, making these kinds of changes to the harvested natural product is often laborious – where it’s possible at all.
Although the usefulness of total synthesis is constantly debated, it’s generally agreed that probing the structure-activity relationship of a bioactive natural product is perhaps one of the few areas where it remains indispensable. But even though several impressive total syntheses of pleuromutilin have been achieved, no synthetic route had been designed with this goal in mind. A modular approach by Seth Herzon and co-workers at Yale University in the US has now set the stage for a detailed investigation of new analogues inaccessible from the natural product itself.2
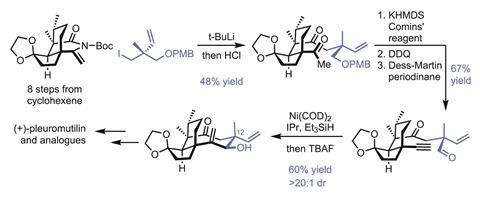
The group begins by quickly assembling the target’s five- and six-membered rings, needing just eight steps to prepare the enantiopure cyclic enimide from cyclohexenone, in 20% overall yield. Next comes the key fragment coupling that installs the remaining half of the cyclooctane ring (figure 2). Adding this unit last – with the flexibility to change its makeup – is really the genius of this approach, and the group will no doubt exploit this modularity in the future to further explore the related chemical space.
Stapling on the remaining carbon atoms begins with an organolithium addition to the enimide. Although the 48% yield seems modest at first, it’s worth noting that both the nucleophile and electrophile are neopentyl, and bringing together two quaternary centres like this is hard. Next, the methyl ketone is converted to the alkyne, and the p-methoxybenzyl-protected alcohol is exposed and oxidised. An exo-selective reductive cyclisation under nickel catalysis completes the final ring. The regio- and diastereoselectivity here are quite impressive, and if this approach can make an eight-membered ring, it’s probably good for other sizes as well, as long as the exo-selectivity holds up. The carbon skeleton is now complete, and just a few oxidation-level tweaks and inversion of the C-12 centre are needed to complete the natural product.
Often to obtain this level of flexibility, chemists are forced to sacrifice efficiency, and while it’s not a manufacturing route, I think this synthesis does a great job of balancing the two. With the group now poised to make libraries of synthetic pleuromutilins, I am excited to see what they find!
References
1 D J Newman and G M Cragg, J. Nat. Prod., 2016, 79, 629 (DOI: 10.1021/acs.jnatprod.5b01055)
2 S K Murphy, M Zeng and S B Herzon, Science, 2017, 356, 956 (DOI: 10.1126/science.aan0003)












No comments yet