
From a seemingly impossible problem a few years ago, some researchers think that predicting the folded structures of protein could be solved pretty soon. James Mitchell Crow reports
Every two years, the international community of protein structure researchers runs a competition. Known as the Critical Assessment of protein Structure Prediction (Casp), it assesses the latest progress in computer modelling to predict the 3D structure of proteins from their amino acid sequence.
For many rounds of Casp, the results were sobering. Researchers were getting pretty good at homology modelling, where they would model a protein’s structure based on that of a closely related protein with a known structure. But when it came to predicting protein structure from amino acid sequence only, things were at a standstill. ‘We were on the brink of abandoning the prediction category because there was absolutely no progress over many Casps,’ says Krzysztof Fidelis, director of the Protein Structure Prediction Center at the University of California, Davis in the US, who has been on the Casp organising committee since the competition’s inception in 1994.
But in 2014, at Casp 11, something changed. ‘Our free modelling assessor, [University of Texas protein modeller] Nick Grishin, wrote to us that someone either was cheating, or had solved the folding problem!’ Fidelis says. ‘It turned out to be neither, but nevertheless – it was an amazing revelation.’
The model that caused Grishin to fire off his email came from David Baker and his team at the University of Washington in Seattle, US. The remarkable strides forward Baker has recently made in predicting protein structure – and in using that ability to design new proteins with specific applications ranging from medicine to catalysis – earned him a runner up spot in Science’s Breakthrough of the Year list for 2016.
Baker’s progress, and that of the wider community, shows no sign of slowing.
Search space
More than half a century ago, protein researchers realised it was a protein’s amino acid sequence that determines the 3D structures into which these long chain molecules fold themselves up. Warm up a protein, and its tightly folded structure will unravel. But allow the protein to cool, and it will coil back up again into its original folded shape as its amino acids arrange themselves back into their lowest energy state.

It is in their folded state that proteins become functional. In the human body alone, those roles range from marshalling the synthesis of complex molecules to converting chemical energy into mechanical work in the muscles, to sensing light in the eye and to sensing traces of airborne small molecules in the nose.
But while experimentally reading off a protein’s amino acid sequence is quick, experimentally determining its folded structure – using techniques such as x-ray crystallography and advanced NMR spectroscopy – remains slow and costly. As just one example of the lengths researchers sometimes have to go to obtain a protein structure, Japanese drug discovery company PeptiDream recently partnered with Japanese space agency JAXA to send protein samples into space, to grow better protein crystals under low gravity, for crystallographic analysis once the sample was returned to Earth.
At the start of 2018, the Protein Data Bank, the international repository of experimentally obtained protein structures, contained the structure of 138,464 proteins. That’s a drop in the ocean compared to the 1012 unique protein structures the natural world is estimated to produce. To fill in some of the blanks, many research groups have tried to develop computational models that can analyse a protein’s amino acid sequence, and calculate how it must fold.
‘The basic principle is that proteins fold to their lowest energy state, just like any physical system – like a ball rolling on a bumpy surface, for instance,’ Baker explains. ‘So the problem of protein structure prediction is the problem of finding the lowest energy state for a protein given its amino acid sequence.’
That might sound simple, but the reality is anything but. The first challenge is to program an energy function that calculates the energy of a folded protein. Then the program has to chug away, testing folded state after folded state, hunting for the one with the lowest energy.
‘Protein structure prediction has been a hard problem because a lot of things can go wrong,’ says William DeGrado, a protein structure modeller at the University of California, San Francisco, in the US. ‘If you think about the number of rotatable bonds, each can do its own thing. It’s only when it all happens together, cooperatively, that you get the right answer,’ he says.
Begin at the beginning…
If a protein closely related to the one in question has previously had its structure experimentally determined, that gives the computer model a huge leg up, a template from which to work from. But for most proteins, there’s no known structure to build a homology model from. The computer model must start from the beginning – ab initio, to use the common Latin phrase – exploring a vast search space to try to find the lowest energy folded structure.
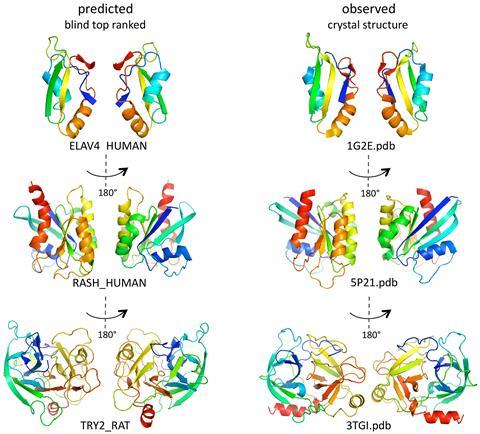
In the first Casp rounds, held in the mid-1990s, ‘ab initio modelling was essentially a non-starter’, Fidelis says. ‘Around Casp 4, in 2000, there was a bit of a breakthrough. People started to be able to fold small proteins, occasionally, with some correctness.’
Baker was one of the pioneers at that time, with his Rosetta software. Rosetta was not an instant success, Fidelis recalls. ‘We looked at their models with some degree of amazement when they were first submitted – they were very extended and un-protein-like,’ he says. But things rapidly improved, and by Casp 4 it was starting to produce some impressive results. ‘David Baker’s group, with Rosetta, were able to fold some of these proteins early on.’
‘When we first developed Rosetta, at the time my lab was really focussed on experimental studies of protein folding,’ Baker says. ‘So we got some intuition about how proteins fold, and those intuitions we built into the basis for Rosetta.’
Three key things have come together to gradually improve Rosetta’s performance over time – not just directly within Baker’s research group, but the international community of collaborators, including many ex-Baker lab researchers, that has grown up around Rosetta.
The first is the gradual improvements this community has made to Rosetta’s energy function, making the program progressively more accurate at calculating a folded protein’s energy. ‘Second, we have better algorithms for searching the structure space,’ Baker says. ‘And third, there is more computing power available today than we had before.’ In particular, the team established the highly popular Rosetta@home – where Baker extended the Rosetta community to include non-scientists. Over a million members of the general public installed a program on their home computer to start solving protein structures whenever the computer was idle.
And then there’s Baker himself, DeGrado says. ‘The guy’s smart as hell. The scale of his operation is immense. The funding and the people that he attracts are absolutely first rate. He’s created this community that works pretty cooperatively. It’s quite a feat, on several levels.’
And yet – until that big Casp 11 breakthrough in 2014 – progress at predicting protein structure was agonisingly slow for all but the smallest proteins. ‘The problem is, as sequences get longer, you never in practice get close to the actual structure,’ Baker says. The search space is simply too large. ‘You could ask, is the bigger problem that your ability to compute energy isn’t accurate, or is it that you never really stumble upon the correct structure?’
Enhanced by evolution
It was Chris Sander, a computational biologist at Harvard Medical School in the US, who first realised that nature – specifically, evolution – might offer clues that could dramatically narrow this search space.1
By comparing the amino acid sequences of large numbers of closely related proteins, especially proteins of the same family from different species, it was possible to identify pairs of amino acids that, however far apart they are along the protein backbone, evolve together. Whenever one changes, so does the other. The fact these amino acids co-evolve suggests they must play a role, as a pair, in the protein’s function – and so are probably found close together in space within the folded protein.
Even if a protein family has no known structures from which to create homology models, as long as enough of the family’s protein sequences are known, identifying co-evolved pairs of amino acids effectively pins together parts of the protein’s folded structure and dramatically reduces the search space the protein folding model must explore.
When Baker and his team plugged this approach into Rosetta – which they did for the first time in time for Casp11 – the improvement was dramatic. ‘We published a paper in Science in 2017 showing that by using metagenome sequence information, we could drastically improve the power of protein structure prediction – to the point you can reliably compute the structures of proteins whose structures currently aren’t known ,’ Baker explains.2 ‘With this coevolution information, it basically makes a big protein more like a small protein: it constrains the search, so a 250-residue protein becomes more like a 60-residue structure, because you have an idea of what space you need to be looking in.’
Fidelis is optimistic that, for most proteins, we’ll soon be able to declare protein folding solved. ‘For the last 20-something years, we have been saying that the protein folding problem will be solved in about five years. I still think this is true!’ he says. This time, things are really coming together, he says. ‘In terms of how the pieces of main chain are arranged in the protein core for a globular protein, I think we will get to there within five years.’
Dare to design
Using the latest co-evolution guided iteration of Rosetta, Baker’s group is working through all the large protein families predicting their structures. ‘For us the interesting thing now is to apply protein prediction methods to build models of proteins that people care about on a large scale,’ Baker says. ‘Our main problem now is how to get them out and available to people, because the PDB considers them models that they don’t want to deposit.’
But most of Baker’s energy these days goes in a new direction. ‘The other problem we work on is protein design. By historical accident, we have life and we have the proteins that exist in nature. But now we understand the principles [of protein folding] we should be able to design proteins to solve current problems.’ When looking for a new protein-based medicine, or a new catalyst, why limit ourselves to the 1012 protein structures nature has stumbled upon? For a typical protein, 200 amino acid residues in length, there are a staggering 20200 possible structures to explore, Baker says.
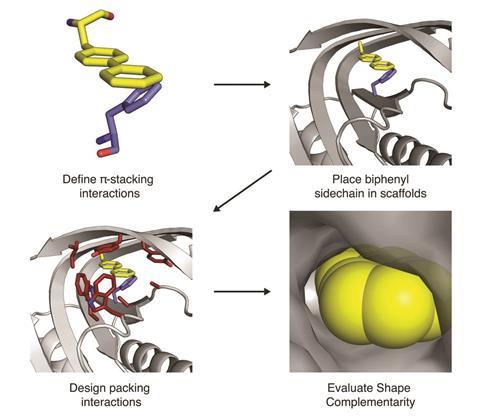
How do you go from predicting the structure of existing proteins to designing new ones? ‘It’s the same thing, except instead of starting with a naturally occurring sequence that came from the genome of some creature, you start at the other end. You start with a brand new structure that you devise on the computer to solve the problem at hand, then you have to find the sequence whose lowest energy state is that structure.’
Perhaps the most straightforward design challenge – relatively speaking – is to design a protein or peptide that simply sticks to its target, with high selectivity and high affinity. The obvious application in this space is medicine. Peptides and small proteins could fit neatly between the traditional small-molecule drugs, and the antibody-based ‘biologic’ drugs that have recently come to dominate lists of blockbuster drug sales.
Small molecule drugs have the advantage that they are relatively easy to make in large quantities, they can be taken in pill form rather than have to be injected, and have a good shelf life. Antibodies are the opposite – but make up for these drawbacks by binding their target much more specifically than small molecules can, improving their potency while reducing side effects.
‘We think [that with] designed molecules in the size between small molecules and antibodies you can get the best of both worlds – things which still bind with very high affinity and specificity to their targets but are easier to manufacture and much more stable,’ Baker says. ‘We’re now trying to figure out what the rules are to try to get these proteins or peptides to cross membranes,’ he adds. ‘Then you really could have all the advantages of the small molecule but still with the higher specificity and affinity. We’re obviously not there yet but that’s what we’re aiming towards.’
Baker’s group is already taking some interesting steps forward. The drug cyclosporine, used as an immunosuppressant for some autoimmune diseases as well as to prevent organ transplant rejection, is a macrocyclic peptide 11 amino acids in size. In November 2017, Baker and colleagues published a comprehensive analysis of the stable structures that can be formed from peptide macrocycles between seven and 10 amino acid residues in size.3 The team identified more than 200 stable structures that could be investigated as scaffolds for future drugs.
But that publication has a deeper significance, Baker argues. ‘People could always say, “Well, the proteins you’ve designed are always made out of alpha helices and beta sheets so how do you know you are not just copying what’s in nature?” You can see when you look at the paper, the things we are designing look nothing like anything in nature, but their design is done in exactly the same way,’ he says. ‘We’re really excited because this shows we can take the principles of what we’ve learned from designing proteins and now apply them to make things that look completely different from what nature’s come up with.’
In another landmark 2017 paper, Baker’s group began to exploit the fact proteins are now not only quick to design.4 It is also quick and inexpensive to synthesise the DNA corresponding to that protein sequence, then transfer that DNA into E. coli and have the bacteria produce the protein; and it also quick to experimentally test the resulting proteins against their target in a high-throughput screen. In this particular example, they designed and tested 22,660 mini-proteins of 37–43 residues that target influenza haemagglutinin and botulinum neurotoxin B.
‘Biology, generally, is a descriptive science – you don’t usually get a chance to develop hypotheses and then to test them rigorously by creating new lifeforms,’ Baker says. ‘But in this case you really can do that. You can have some hypothesis about what type of protein would bind the flu, and then you can test 10,000 or 100,000 hypotheses to figure out which ones have the most validity. And then keep iterating,’ Baker says. It’s a positive feedback loop that allows the very process of protein design to be improved, potentially springboarding big advances – in the same way the co-evolution data recently did for protein structure prediction. ‘It’s very exciting to be able to collect data on your computational model at that scale,’ Baker says.
Creating catalysts
The next area Baker is focusing on – or rather, refocusing on – is protein catalysis.
That’s a much tricker proposition than simply binding a target, says DeGrado. ‘With an enzyme, one needs to bind to the ground state, and one needs to bind to the transition state and any other high energy intermediate, and then one needs to bind weakly to the products. It’s a multi-state design problem,’ he says.
But if we can get there, the potential is vast. Proteins, in the form of enzymes, are exceptional catalysts – despite being limited to a very restricted range of bioavailable metal ions such as iron and nickel. ‘We live in a biological world with a limited number of cofactors for catalysis,’ says DeGrado. ‘But chemists use huge numbers of cofactors including various metal ions.’ The aim is to design proteins that incorporate some of these potent cofactors – things like platinum, palladium and rhodium, to marry the versatility and efficiency and programmability of protein structures with the catalytic capability of manmade cofactors . ‘That’s an interesting area of research I think is going to take off.’
Proteins do so many different things in biology
In 2010, Baker published the design of an enzyme that could catalyse a bimolecular Diels–Alder reaction.5 ‘At that point, we were computing ideal active sites from first principles, and then searching through native protein structures for a place we could embed that active site,’ Baker says. But it was never possible to find a natural protein where the designed active site would perfectly fit, which compromised performance. ‘Now we’re really focused on de novo protein design, we’re building scaffolds from scratch to harbour the designed site.’
Baker’s team is yet to publish any result in this area – but watch this space. As DeGrado recently demonstrated, designing a protein as a whole can reap big rewards.6 A classic problem of protein design has been to design proteins that can selectively bind a small molecule porphyrin, he says. ‘Previously we tended to think of designing the protein ligand binding site, and the rest of the fold of the protein and its hydrophobic core as separate problems.’ But as research into antibodies has shown, regions of the protein that are quite far from the binding site can contribute quite a bit to affinity and specificity, DeGrado says. ‘In a similar manner, we thought we have to design the entire protein, including those portions that might just seem like framework, all in one computation so that it’s all working together and playing well.’ The very first protein they designed this way worked beautifully at binding the porphyrin, he says.
‘We hope that approach will be applicable to other small-molecule binding problems. We’re developing a lot of new code that revolves around that idea,’ DeGrado says. ‘I think it’s going to be a very fruitful area.’
‘Proteins do so many different things in biology,’ Baker says. ‘As we get better at the protein design, hand in hand with that we’re able to design more and more sophisticated things.’
James Mitchell Crow is a science writer based in Melbourne, Australia
Misfolded medicines
Think of the word ‘amyloid’, and the chances are neurodegenerative diseases such as Alzheimer’s and Parkinson’s will spring to mind. Amyloids are aggregates of misfolded proteins that characteristically form in the brains of neurodegenerative patients, appearing to physically clog the brain. Once these misfolded proteins begin to form, a runaway process starts to occur where these misfolded clumps seed the misfolding of any other protein they come into contact with.
Frederic Rousseau and Joost Schymkowitz from the University of Leuven, Belgium, are taking the idea of amyloids and turning it on its head. They’re designing amyloids that treat disease, not trigger it. They are designing proteins that will seed the misfolding and aggregation of proteins found in cancer cells and pathogenic bacteria.
The work began after the team designed a program that could identify aggregation-prone stretches of amino acid sequence in proteins. ‘Then we developed this algorithm, Tango, and we started doing it for analysing whole proteomes,’ Schymkowitz says. The results were a shock. ‘Very soon we realised that most proteins, if not all, have aggregation-prone regions.’
Only a small proportion of the proteins in the human body have ever been known to form amyloids – but it turns out virtually every protein has the potential for it. That seeded the idea that deliberately triggering protein misfolding could be a way to treat disease.
In a paper in Science in 2016ac, the team designed a peptide that could successfully penetrate cancer cells and trigger the formation of protein aggregates of its target protein, VEGFR2, a protein essential for the survival of certain cancer cell types.7
‘We’re exploring different targets,’ Schymkowitz says. ‘We are looking at other targets now to see if we could find things where a small molecule struggles or where antibodies can’t reach. Cancer is definitely a focus, but also antimicrobials.’
One particularly promising area is to hit multi-drug-resistant bacteria. ‘What we see so far is that resistance to beta-lactam antibiotics doesn’t correlate in any way to resistance against aggregating peptides. Hopefully it could provide a backup, which we will badly need if antibiotic resistance keeps rising as it is.’
References
1 D S Marks et al, PLoS ONE, 2011, 6, e28766 (DOI: 10.1371/journal.pone.0028766)
2 S Ovchinnikov et al, Science, 2017, 355, 294 (DOI: 10.1126/science.aah4043)
3 Hosseinzadeh et al, Science, 2017, 358, 1461 (DOI: 10.1126/science.aap7577)
4 A Chevalier et al, Nature, 2017, 550, 74 (DOI: 10.1038/nature23912)
5 J B Siegel et al, Science, 2010, 329, 309 (DOI: 10.1126/science.1190239)
6 N F Polizzi et al, Nat. Chem., 2017, 9, 1157 (DOI: 10.1038/nchem.2846)
7 R Gallardo et al, Science, 2016, 354, aah4949 (DOI: 10.1126/science.aah4949)











No comments yet