

Super cool microscopy wins the 2017 Nobel prize in chemistry
‘It was a very confusing experience,’ Joachim Frank told a press conference at Columbia University in the US, recalling the moment he found out he had been awarded a share in the 2017 Nobel prize for chemistry. ‘I was sure I was going to be woken up by my new dog, but then this phone call arrived.’
At around the same time Frank was being woken up, Richard Henderson from the Medical Research Council Laboratory of Molecular Biology (LMB) in Cambridge, UK, was listening to a talk at an electron microscopy conference. Then his phone rang. ‘I thought “I’m in a meeting surrounded by people, I can’t talk now”, so I rejected the call,’ he says. But when he noticed that the call was coming from Sweden, he left to return the call. ‘Meindert [Lamers, another LMB researcher] thought I was being a bit rude by going out,’ Henderson laughs.

Alongside Jacques Dubochet from the University of Lausanne, Switzerland, Frank and Henderson were honoured for their work ‘developing cryo-electron microscopy (cryo-EM) for the high-resolution structure determination of biomolecules in solution’.
Cryo-EM lets scientists look at the machinery of life – DNA, ribosomes, proteins and viruses – in its most lifelike state. The cool microscope can decipher almost any biomolecule’s structure. After many years of standing in the shadow of methods such as x-ray crystallography, cryo-EM is now one of the fastest growing areas in structural biology.
But it’s been a long road. The laureates’ belief in cryo-EM’s potential, and their determination to overcome its challenges, laid the groundwork over decades to eventually deliver a method that now makes it easier than ever before to capture biological structures in atomic detail. ‘[It’s] a well-deserved prize, a long time coming,’ says Kirsty MacLellan-Gibson, a former postdoc in Dubochet’s group and now head of biological imaging at the National Institute for Biological Standards and Control in South Mimms, UK.
From x to e
Since the 1950s, when scientists first started looking at life’s molecular structures, x-ray crystallography has been the method of choice. Today, 22 Nobel prizes have been awarded to structures elucidated by x-ray, including DNA, myoglobin, ion channels and receptors.
However, as its name suggests, it relies on having a crystal: the regular arrangement of atoms in a crystalline solid. Biomolecules aren’t rigid by nature, which often makes them tricky or impossible to crystallise. And in forming dry crystals, proteins can be forced into shapes they wouldn’t adopt naturally. Cryo-EM on the other hand, can work without a 3D crystal and retains one of life’s most essential components: water.

In the early 1970s, Henderson was struggling to make crystals of bacteriorhodopsin, a proton pump protein that sits in cell membranes of some bacteria. Isolated from the membrane, the protein simply lost its structure, and no crystal meant no x-ray crystallography.
Then in 1973, he recalls ‘I heard a talk by Nigel Unwin about his efforts to improve electron microscopy’. With the wavelength of electrons just right to interrogate atomic details, and capable of imaging thin 2D samples that x-rays would simply pass straight through, EM sounded like the perfect method for his protein.
Invented in the 1930s – and netting a Nobel for Ernst Ruska in 1987 – EM had been extensively used to study pigments, paints, metals and other non-living matter. But the electron microscope isn’t very amenable to biomolecules: it requires a high vacuum to work (so that the electron beam isn’t scattered by gas molecules), which dries the samples out, and the energy of the electron beam destroys covalent bonds. ‘You will basically burn away the entire molecule,’ explains Sjors Scheres, Henderson’s colleague at LMB.
Lower electron doses can limit the damage, but that means a weaker contrast. One solution at the time was to use negative stains – applying heavy metal salts to a sample. ‘The stain forms a shell around the outside of the protein,’ explains MacLellan-Gibson. ‘You get really high contrast at the outside, but it masks the internal structures because the electrons are interacting with the atoms [around the protein] rather than the atoms in the sample,’ she continues.
Henderson and Unwin were unsatisfied with this approach; they wanted to map bacteriorhodopsin’s internal structure. They prepared a lipid bilayer membrane containing bacteriorhodopsin molecules and replaced the water with a glucose solution to keep it from drying out. A weak electron beam helped to minimise structural damage.
The team also took advantage of bacteriorhodopsin’s unusual feature: it is effectively a 2D crystal, naturally arranged inside the membrane in a highly regular manner. This means that any damage to an individual protein was negligible when averaged out over the whole sample.
In 1975, bacteriorhodopsin with its seven helical protein strands became the first biomolecule structure to be solved using electron microscopy.1 However, with a resolution of 7Å, there was still some way to go to reach x-ray crystallography’s 3Å resolution. It would be another 15 years before Henderson obtained bacteriorhodopsin’s structure in atomic detail.2
A different angle
While Henderson’s breakthrough had taken advantage of the protein’s periodic structure, other samples contain randomly scattered biomolecules – all in different orientations.
In the early days of EM, scientists like crystallographer Walter Hoppe at the Max Planck institute in Munich, Germany, tried to overcome this problem using tomography. This means reconstructing a 3D structure by taking snapshots of the sample from different directions. Frank was Hoppe’s PhD student, and he was critical of his mentor’s method: ‘He approached this idea from an angle that I thought was not effective or was simply wrong,’ Frank says.
Tomography’s requirement for multiple images means the sample gets bombarded with high electron doses numerous times, Frank explains. ‘In the end, you have exposed a molecule to such an extent that the result is meaningless. If you form a 3D image from something like this, you might as well forget it.’
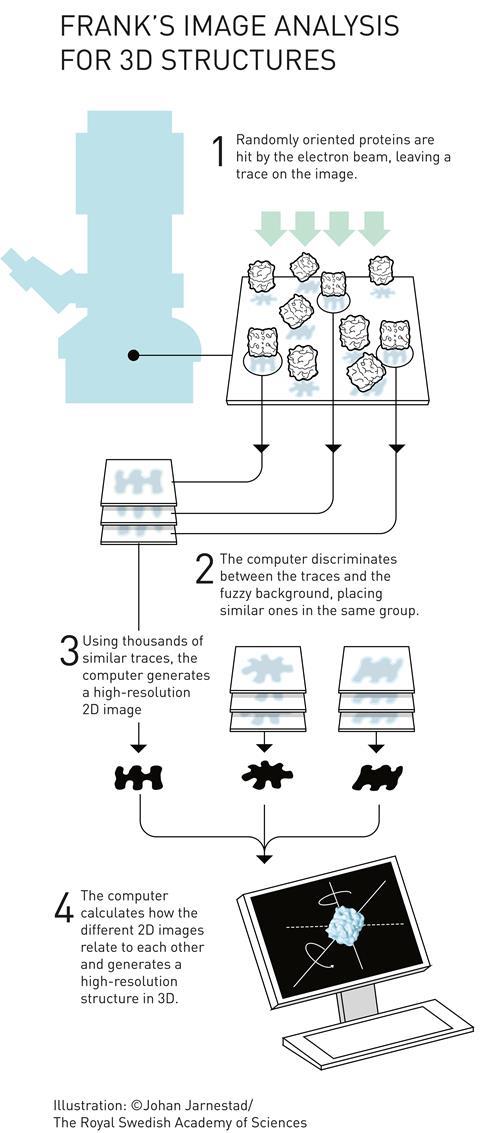
Frank realised that with millions of molecules per sample, a single snapshot already contains many different orientations. ‘So you don’t have to tilt the molecule,’ he continues. ‘It tilts itself, that’s the idea.’ ‘Each of those images is extremely noisy’, explains Scheres. ‘But provided you have enough images, you can actually align and average them. By averaging, the signal remains but the noise gets averaged away.’
In the early 1980s, Frank and Marin van Heel, then visiting student in Frank’s lab and now researcher at the National Nanotechnology Laboratory in Brazil, published a statistical computational method that groups and averages molecules in different orientations and states.3 However, a problem remained: how do the 2D images relate to a 3D structure?
Reconstructing reality
‘The image that you obtain is actually a 2D projection,’ explains Henderson’s LMB colleague Lori Passmore. ‘That means that all of the three-dimensional information is collapsed down into two dimensions; even the inner structures,’ she continues.
Frank reconstructed 3D images using the random conical tilt method.4 He took two images, one with the sample lying perpendicular to the electron beam and one in which the sample is tilted. This allowed him to determine each particle’s angle towards the electron beam. A mathematical trick in the form of forward and reverse Fourier transformation then revealed the original 3D object (see below).
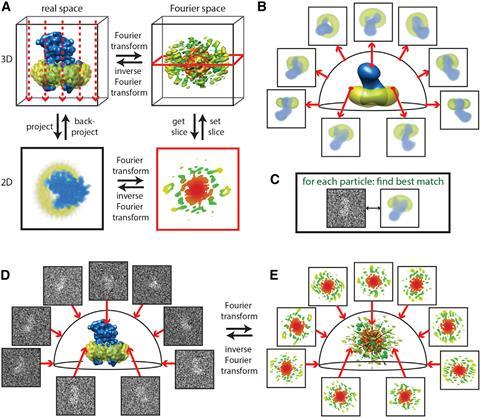
‘A two-dimensional Fourier transform of the projection is a central section through the 3D Fourier transform of the original object,’ Scheres elaborates. ‘By having multiple 2D projection images from different directions, if you know the relative directions, you can put all those 2D slices back and do the reconstruction through inverse Fourier analysis.’
Frank built a computer program called Spider (system for processing of image data in electron microscopy and related fields) to do tilt angle grouping and 3D reconstruction.5 He demonstrated its power in 1987 when he used it to reconstruct part of a bacterial ribosome,6 and it has remained a favourite with microscopists over the years. Only recently it has been overtaken by other programs, such as Scheres’ Relion (regularised likelihood optimisation), which require less programming expertise.
Liquid ice
At the same time as Frank was working on Spider, Dubochet had just started his own research group at the European Molecular Biology Laboratory in Heidelberg, Germany. Dubochet wasn’t happy using specimens that were dried, or had their water replaced with glucose or plastic resins. He wanted to see biomolecules in their natural, hydrated state. ‘If you have it without water, the molecules, like fish, are dead,’ Dubochet said in an interview with Nobel media.
‘He had a love of water from the beginning,’ recalls Alasdair McDowall, director the EM centre at California Institute of Technology, US, who worked with Dubochet at the time. ‘Water is beautiful in his eyes. It has to be because that’s what life is all about.’
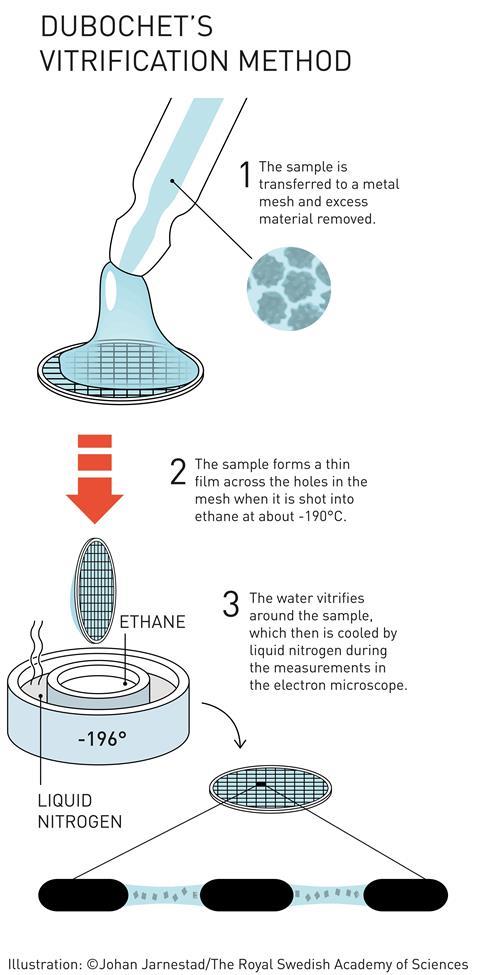
Attempts had been made previously to use cooling to stop samples dehydrating in the microscope’s vacuum, as well as to protect them from radiation damage. The problem is that cold water makes ice. And apart from destroying delicate biological structures, crystalline ice strongly diffracts the electron beam. Most EM images taken from frozen samples were useless.
But if the ice isn’t crystalline, then it is almost completely transparent to electrons. Dubochet’s team therefore worked on embedding samples in vitreous ice – an amorphous, glass-like form of frozen water. Up to that point, vitreous ice was rare - only observed as thin layers on supercooled metals. The idea that water droplets could be frozen into vitreous ice was considered bold to say the least.
That didn’t deter Dubochet. Using a homemade plunge freezer (essentially a retort stand with a few elastic bands), his team started looking for a liquid that could cool water so quickly it became vitreous. ‘We propelled samples into different cryogens,’ McDowall explains. ‘We went through the whole chemistry book of organics for cryogens that would stay liquid at the lowest temperatures possible,’ he recalls.
‘And then, one day, everything changed with what I call an “aha” moment, a unique moment when one understands that one has just made a real discovery,’ Dubochet told the Swiss daily newspaper Le Temps. His team had found that ethane, cooled to –190°C by liquid nitrogen, was perfect.
His group finally published the first working plunge freeze setup, consisting of a nebuliser and a cold bath. The sample holder was shot through a spray of water droplets directly into the cryogen, from which it was then extracted and placed in an equally cold microscope.7
Although Dubochet noted that ‘we still do not understand the nature of water vitrified by our method’, his method transformed EM to cryo-EM. The vitrified water became the last piece in a methodology puzzle that gave researchers for the first time access to clear images of unstained biomolecules.
Atoms from blobs
By the early 1990s, cryo-EM had gained a small scientific following, but ‘it was still difficult and people were making more progress in understanding biology by using crystallography, or by combining the two things,’ Passmore explains. Getting to atomic resolution seemed like an insurmountable obstacle. ‘We were stuck at around five to seven angstroms,’ Frank says.
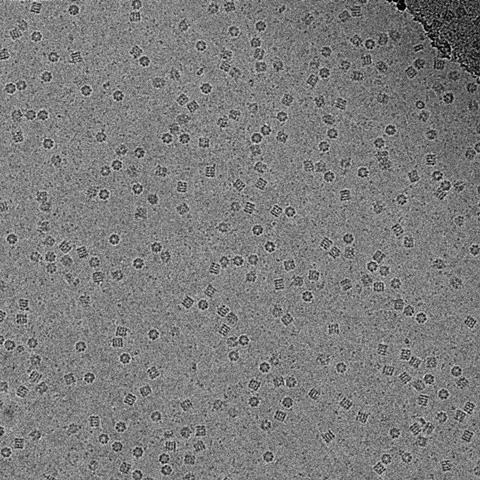
It became known as the ‘blobology era’, Henderson remembers. ‘You did a structure and you got blobs – you can’t see what the atoms are, no chemistry yet.’ But he was determined not to let go of his dream, even though few shared his vision. ‘Let’s say, many people were thinking he was a bit of an optimist,’ Scheres laughs.
Resolving the blobs relied on another factor. At the time, and until around 10 years ago, electron microscopists still used photographic film to capture their images, despite having to develop and scan each individual photo – the digital sensors available didn’t capture electrons directly, instead converting the electron signal into a light signal, which adds noise. However, photographic film only captures about a quarter of the electrons. ‘Because you have to be so careful with how many electrons you use, that yielded very noisy images,’ Scheres explains.
So when direct electron detectors came to the market in 2012, cryo-EM was ready. Structures resolved to below 3Å – previously the realm of x-ray crystallography – aren’t unusual anymore, says Bridget Carragher from the is New York Structural Biology Center in the US. In 2015, her team resolved the structure of a protein-degrading complex to 2.8Å,8 and a year later another group resolved a protein structure to 1.8Å.9
One of the smallest structures ever imaged is human γ-secretase, a membrane enzyme that generates the short amino acid filaments that aggregate in the brains of Alzheimer’s patients.10 And during the 2016 Zika virus epidemic, researchers managed to image the virus to 3.8Å resolution in just a few months.11

Cryo victory
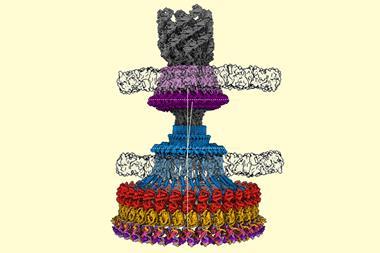
More than four decades after Henderson first worked with EM, ‘you can now put more or less anything in the microscope: single particles, viruses, 2D crystals, 3D crystals, bits of cells, organelles,’ he says. Cryo-EM has also evolved to include a fourth dimension: time. Instant freezing preserves the molecules in their different dynamic states, which gives scientist the unique opportunity to observe the cell’s molecular machinery in action. There are movies of the human ribosome turning genetic code into proteins12 and of ATP synthase, a molecular turbine converting a proton flow into the cell’s energy currency adenosine triphosphate.13
The number of cryo-EM derived structures being deposited in the Protein Data Bank is increasing each year: in 2010, just 53 structures were deposited; in 2016 it was over 400. ‘Cryo-EM has really exploded in the last five years,’ Passmore says. ‘It’s rare that you see a field move so quickly, changing right before your eyes.’
And EM scientists are setting the bar ever higher. ‘Looking at structures in situ is one of the remaining holy grails of EM,’ says Carragher. The way forward could be focused ion beam milling, which uses a scanning electron microscope to cut away sections of a frozen samples until only a small wedge remains. ‘This is going to be a big opening for understanding where proteins are going, moving and all the activities of viruses within cells that are often deep in parts we can’t see,’ McDowall says. Carragher adds: ‘This work is still very challenging but enormously exciting and we are all looking forward to new technical developments in this area.’
Einstein from noise
Although sophisticated computational tools can extract beautiful 3D structures from low resolution EM images, they have their pitfalls. In 2013, Harvard University researchers claimed to have elucidated the structure of a key HIV proteina – a claim that was strongly disputed by Henderson,b van Heelc and Sriram Subramaniamd from the Center for Cancer Research, Bethesda, US.
The three scientists argued that the Harvard team fell for a the ‘Einstein from noise’ trap: finding images in random background noise. ‘You can find anything you want in random white noise,’ van Heel explains. Automated particle search – particularly when done with the computer being given a reference structure – makes it possible to find anything, even if the sample doesn’t contain any particles at all.
The Harvard team countered the criticism, detailing how their image extraction method was specifically designed to avoid the problem. Their resolution of 6Å, they argued, could not have been attained using a previous structure with 11Å resolution as reference. The dispute remains unresolved.
Today’s direct electron detectors are so sensitive that scientists can see structures with the naked eye and become less reliant on computers to distinguish molecules from noise. ‘We’re now doing things completely reference-free,’ says van Heel, although he cautions that computer programs and their users are still prone to bias that can make an Einstein out of noise.e
References
1 R Henderson and P N T Unwin, Nature, 1975, 257, 28 (DOI: 10.1038/257028a0)
2 R Henderson et al, J. Mol. Biol., 1990, 213, 899 (DOI: 10.1016/S0022-2836(05)80271-2)
3 M van Heel J and Frank, Ultramicroscopy, 1981, 6, 187 (DOI: 10.1016/0304-3991(81)90059-0)
4 M Radermacher et al, J. Microsc., 1987, 146, 113 (DOI: 10.1111/j.1365-2818.1987.tb01333.x)
5 J Frank, B Shimkin and H Dowse, Ultramicroscopy, 1981, 6, 343 (DOI: 10.1016/S0304-3991(81)80236-7)
6 M Radermacher et al, J. Microsc., 1987, 146, 113 (DOI: 10.1111/j.1365-2818.1987.tb01333.x)
7 J Dubochet and A W McDowall, J. Microsc., 1981, 124, 3 (DOI: 10.1111/j.1365-2818.1981.tb02483.x)
8 M G Campbell et al, eLife, 2015, 4, e06380 (DOI: 10.7554/eLife.06380)
9 A Merk et al, Cell, 2016, 165, 1698 (DOI: 10.1016/j.cell.2016.05.040)
10 X-c Bai et al, Nature, 2015, 525, 212 (DOI: 10.1038/nature14892)
11 D Sirohi et al, Science, 2016, 352, 467 (DOI: 10.1126/science.aaf5316)
12 E Behrmann et al, Cell, 2015, 161, 845 (DOI: 10.1016/j.cell.2015.03.052)
13 J Zhao, S Benlekbir and J L Rubinstein, Nature, 2015, 521, 241 (DOI: 10.1038/nature14365)
a Y Mao et al, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 12438 (DOI: 10.1073/pnas.1307382110)
b R Henderson, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 18037 (DOI: 10.1073/pnas.1314449110)
c M van Heel, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, E4175 (DOI: 10.1073/pnas.1314353110)
d S Subramaniam, Proc. Natl. Acad. Sci. U.S.A., 2013, 110 E4172 (DOI: 10.1073/pnas.1313802110)
e P Afanasyev et al, IUCrJ, 2017, 4, 678 (DOI: 10.1107/S2052252517010922)












No comments yet