
Industry and academia are turning to antimicrobial peptides to find new antibiotics, Andy Extance discovers
In the soil of Mount Ararat, Turkey, live the bacteria that yielded the last truly new class of antibiotics.1 Scooped up and sent to the labs of pharmaceutical giant Eli Lilly in Indianapolis, US, the compounds the bugs made were developed through the 1980s and 1990s. But when the resulting drug candidate – a lipopeptide with the generic name daptomycin (tradename Cubicin) – reached clinical trials, the intravenous dose that was needed to effectively treat infection also damaged patients’ muscles. Supported by Lilly’s then vice-president for infectious diseases discovery, Barry Eisenstein, the company shelved the drug. Then, in the mid-1990s, it stopped natural product antibiotic research altogether, with many researchers leaving the company – including Eisenstein – and daptomycin left in limbo.
At the same time, similar stories were unfolding across the whole pharmaceutical industry, with leading companies cutting both natural product and antibiotic research. But as Eisenstein and his colleagues went on to show, in doing so they abandoned powerful potential ammunition for the battle between bacteria and people. Now, the World Health Organization is warning of a ‘post-antibiotic era, in which common infections and minor injuries, which have been treatable for decades, can once again kill’. With such warnings evoking biblical prophecies, it’s appropriate that interest is again surging in compounds similar to the one found on Ararat, where the Old Testament says Noah’s ark landed. Scientists are turning to peptides to help hold off a flood of resistant bacteria.
Peptide chains have featured in antibiotics since the boom years when discoveries regularly sprung from the soil. Although vancomycin, whose structure combines amino acids and sugar rings, was discovered in 1953, we didn’t discover how important unmodified peptide chains were for fighting bacteria until 1980. Since they were first isolated in the cecropia moth, antimicrobial peptides (AMPs) have been found in all multicellular organisms, where they play a key role in the innate immune system. ‘Why do insects do well without antibodies and lymphocytes?’ asks Paul Hansen from the University of Copenhagen in Denmark. ‘Because they have antimicrobial peptides.’
Breaking down cell walls
AMPs often kill both Gram-negative and Gram-positive bacteria rapidly. That’s important because a negative Gram stain result indicates high levels of the endotoxin lipopolysaccharide in bacterial cell walls, which stimulates the immune response responsible for fever and other symptoms. No new antibiotic class effective against Gram-negative species has been discovered since the fluoroquinolones in the 1960s. Equally critical, given that resistance to some newer drugs has emerged shortly after they reached the market, it’s thought AMPs don’t easily create drug-resistant mutant strains.
AMPs typically associate with bacterial cell walls and either punch holes in them or dive through and attack internal targets. ‘The bacterial membrane is negatively charged, and most antimicrobial peptides are hydrophobic and cationic,’ says Hansen. ‘That means they are electrostatically attracted to the bacterial membrane. An animal cell is typically zwitterionic and contains a lot of cholesterol in the membrane, so that means it’s not nearly as active against them.’
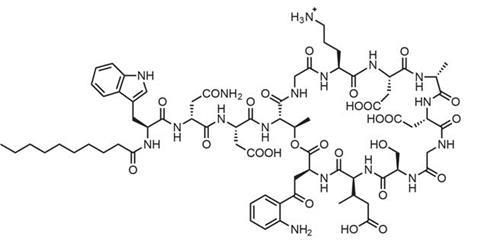
Daptomycin, which is a negatively charged lipopeptide but still punctures bacterial cell membranes, showed that toxicity in human cells remains a concern. However, antibiotic specialist Cubist Pharmaceuticals in Lexington, US, still thought it could be useful if the balance of antibacterial action to muscle damage could be improved. After licensing daptomycin from Eli Lilly in 1997, Cubist pursued one especially important clue in meeting that goal: In animal studies one large single dose, rather than two doses a day, minimised muscle damage.
‘Daptomycin works by aggregating in the presence of calcium ions,’ explains Eisenstein, who’s now Cubist’s senior vice-president for scientific affairs. ‘That produces pores that cause the bacteria to depolarise by loss of internal potassium ions, thereby shutting down DNA, RNA, and protein synthesis. Antibiotic potency is directly related to antibiotic concentration. In contrast, mammalian muscle cells are injured not by concentration but rather by prolonged exposure to the drug without recovery. Thus, less frequent but higher doses simultaneously increase potency and decrease muscle toxicity.’ The US Food and Drug Administration (FDA) approved daptomycin for use in skin and tissue infections in September 2003 and for Staphylococcus aureus in the blood in May 2006.
Back from obscurity
Others are now hoping to follow Cubist’s example of reviving candidates previously dropped during development. For example, Dipexium Pharmaceuticals, in New York, US, was set up specifically to bring Locilex (pexiganan), a 22-amino acid AMP first isolated from the skin of the African clawed frog, to market. Pexiganan is being developed as a cream to treat diabetic foot infection. Dipexium acquired the rights in 2010, after the FDA turned down a new drug application from now-defunct Magainin Pharmaceuticals in 1999.
We’re looking at submitting a new drug application next year
David Luci
‘It was unanimously approved for safety,’ Dipexium chief executive officer David Luci points out. However the FDA called – unfairly in Magainin’s eyes – for another, placebo-controlled, trial, and also had concerns over manufacturing. ‘One was water separation in the cream,’ Luci says. ‘The other was over 5% impurities in the active pharmaceutical ingredient.’ Those issues are connected to a more general manufacturing concern for AMPs: that if they are made chemically rather than by fermentation like daptomycin they’ll be too expensive. Yet Luci denies that the laborious sequence of adding amino-protected residues and then deprotecting them is costly.
‘The peptide is cheap as chips to make,’ he says. ‘We hired Polypeptide Laboratories to manufacture the active pharmaceutical ingredient under today’s manufacturing standards, which are much more developed than 15 years ago. They have the impurities down to 2%. The physical instability issue has also been resolved by DPT Laboratories, a division of Smith and Nephew. All that remains is the Phase III placebo study. We’re now doing those studies and looking at submitting a new drug application amendment in the second half of next year.’ Formulation as a skin cream not only minimises toxic side-effects from pexiganan’s membrane-disrupting mechanism, it also avoids causing systemic resistance, because it has no systemic involvement, Luci explains.
Redirecting garbage disposal
Acyldepsipeptides (ADEPs) are another blast from the past, having first been claimed in a patent application filed by Lilly in 1982. Produced by bacteria collected in an Arizona canyon, Bayer Healthcare revisited them in the 2000s, when Lilly’s patents had expired. ‘The compounds coming out of the bacterium were completely inefficacious in mouse models of infection, so Bayer initiated a medicinal chemistry program,’ says Jason Sello from Brown University in Providence, US.
Bayer found ADEPs kills bacteria differently to AMPs. ‘They bind a peptidase called ClpP, which we think of as garbage disposal for cells,’ Sello says. ‘When proteins are incompletely synthesised and tagged for degradation, they are broken down by ClpP. The ADEPs target ClpP and in binding enable the enzyme to break down unstructured peptides and some proteins that have not been tagged for degradation. Imagine the garbage disposal in your kitchen opening up and chewing on whatever it finds.’
Before Bayer dropped the ADEPs it boosted potency by replacing N-methylalanine with the cyclic amino acid pipecolic acid, stiffening the once floppy peptide ring at the molecule’s heart. That reduces the entropy lost when the ADEP binds ClpP, making the interaction more energetically favourable. Looking for further improvements like this, the Brown team added extra substituents to the pipecolic acid and another key amino acid.
‘The rigidified ADEPs were up to sevenfold better than the natural products binding to ClpP and activating peptidolytic activity in a test tube. However, the changes improved ADEP potency in antibacterial assays by as much as 1200-fold,’ Sello says.2 He credits the leap to the ADEPs being better able to penetrate cells. The stiffening strengthens hydrogen bonds that form between different sides of the ring, reducing hydrogen bond formation with water. Less closely bound to surrounding water, the compounds enter bacterial cells more easily.
Brown University is now working out licensing agreements in the hope of forming an industry–academia partnership. Why would that now be successful, when Lilly and Bayer’s interest waned? Sello admits pharma has moved away from natural product programmes ‘that should exist today’, and suggests companies have also been put off by peptides’ costly reputation. However, he cites Halaven (eribulin), the marine-sponge derived polyketide anticancer drug that Japanese firm Eisai makes via a 62-step synthesis. ‘Halaven is wickedly potent, so the dosing is very small,’ Sello says. ‘Our ADEP is wickedly potent too, so the amount needed might be particularly low.’
Urgent threat
Structural modification has also been important for Cubist, even though daptomycin is made by bacterial fermentation. It’s near to reaping the rewards of focusing on Clostridiumdifficile, which the US Centers for Disease Control and Prevention categorises as an ‘urgent threat’. Following its existing non-peptide C. difficile-associated diarrhea (CDAD) therapy Dificid (fidoxamicin), its investigational oral antibiotic surotomycin, closely related to daptomycin, is in Phase III trials for the same disease.
‘The goal of surotomycin optimisation was to increase the potency of the lipopeptide molecule against the target organism without causing increased potency against the normal gastrointestinal microbiome,’ Eisenstein says. ‘Our analoguing efforts have maintained the daptomycin scaffold and have been pursued either by semisynthesis – chemical modification of the tail and amino acid side chains – or through biosynthetic analoguing (modification of the genes in the producing organism).’
Surotomycin has been granted Qualified Infectious Disease Product (QIDP) status, a recent FDA designation that speeds the review process. Other new incentives, like a potential five year extension of marketing exclusivity, are having an impact, Eisenstein says. ‘In recent years, a more favourable regulatory environment has helped create some traction in rebuilding interest in the area and strengthening the pipeline.’
Molecular Lego

Still, many big pharma firms are absent from antibiotic development. Hansen sees that as a problem, although he understands the fact that new drugs would be probably held in reserve puts them off. His team is therefore seeking to address the serious unmet need for new antibiotics for Gram-negative bacteria like Escherichia coli and Pseudomonas aeruginosa. Rather than revive an old lead molecule, the University of Copenhagen chemists started from anoplin, an AMP discovered in wasp venom in 2001 and just 10 amino acids long.
‘We were looking for small peptides that we could do structure–activity studies on to figure out which amino acids are important,’ Hansen says. His team started by replacing each amino acid with alanine to see how the side chains contribute to its activity. ‘Once you have an idea which ones are important you can play “molecular Lego” to improve the?activity.’
Similarly to other AMPs, anoplin analogues appear to disrupt bacterial cell membranes. ‘The biggest problem is it’s difficult to dissociate from activity against red blood cells,’ Hansen admits. ‘You also want selectivity against certain other pathogens.’ Like Luci and Sello, he doesn’t feel that peptide cost need be an issue, citing Roche’s introduction of the peptide anti-HIV drug Fuzeon (enfuvirtide). ‘They synthesised it in tonne scale and the price of peptide reagents dropped drastically. Maybe the same is going to happen if peptide-based antibiotics take off.’
Another concern peptide drugs face is that they’ll be broken down by peptidase enzymes in the body before they kill enough bacteria. That threat can be minimised by modifying peptides to a form peptidases wouldn’t recognise, as Hansen’s team has shown by producing peptoid anoplin analogues.3 ‘Peptoids move the peptide side chain from the a-carbon to the nitrogen in the backbone,’ he explains. ‘They’re a lot more stable to proteolytic digestion.’
Safe at last
While the journey from university research to peptide-based antibiotics that are a real medical option isn’t easy, Polyphor is now tantalisingly close to showing it can be done. Established in Allschwil, Switzerland, in 1996, Polyphor uses a Protein Epitope Mimetics (PEM) drug discovery platform it developed together with John Robinson at the University of Zurich. PEMs are cyclic molecules, amino acids chains whose ends are joined together by what Polyphor calls a ‘template’, such as a d-proline–l-proline dipeptide. Many AMPs display a similar ß-hairpin structure, so they were a logical starting point for research, explains Daniel Obrecht, Polyphor’s chief scientific officer.
‘Our very efficient parallel synthesis process allows us to rapidly optimise structural and physicochemical properties of PEMs,’ Obrecht says. ‘You can optimise the template, the building blocks, but as you can imagine there are many possible permutations you can do, so you need guided optimisation. It’s a combination of analysing the data and the intuition and knowledge of the medicinal chemists who actually optimise the?molecules.’
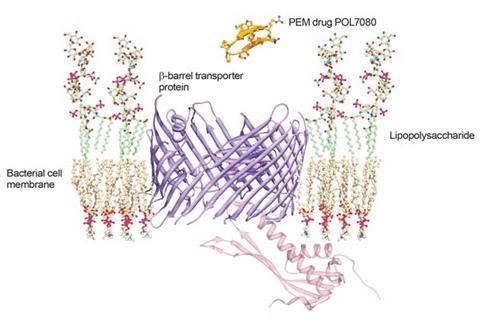
Starting from the AMP protegrin I, Polyphor has produced a lead compound known as POL7080 using this approach. ‘We actually discovered a novel bacteria-specific mechanism distinct from the typical antimicrobial peptides,’ Obrecht says.4 ‘Our molecule isn’t destroying the membrane, but rather targets transport of lipopolysaccharide, which is specific for Gram-negative bacteria. It’s definitely a novel mode of action and therefore doesn’t belong to any other antibiotic class.’
The new mechanism allows POL7080 to escape the toxicity problems that Obrecht says give the ‘hole-punchers’ a figurative ‘ambiguous smell’. Polyphor also says that PEM molecules can be optimised to be stable against proteolytic degradation and produced ‘cost-efficiently by a well-established and scalable synthesis process’. Thanks to these advantages, Polyphor is now partnering with Roche on Phase II trials for intravenous POL7080 administration against P.?aeruginosa?infections.
‘In the discussions with pharma companies it was of prime importance to show that our molecules are different and safe,’ Obrecht says. ‘At the research stage we have further PEM antibiotics that support the claim that POL7080 is not a single compound but actually a new emerging drug class. And we do have clear evidence that you can reach out to other Gram-negative bacteria strains. All these things together show how remarkable this discovery was.’
The determination among scientists and the innovative drug companies at the forefront of peptide antibiotic research show the WHO’s stark warnings are not going unheeded. However, reviving ideas that have previously been linked to toxicity and cast aside may not seem to be the most promising drug discovery model for the long-term. At the very least, AMPs and related molecules are keeping the hope alive that modern medicine will stay afloat despite the rising tide of bacterial resistance. But in fact they’re much more than just a desperate barrel-scraping – they’re a demonstration of how modern science solves problems too hard to crack in the past.
Andy Extance is a science writer based in Exeter, UK
References
1 B I Eisenstein et al, Clin. Infect. Dis., 2010, 50, S10 (DOI: 10.1086/647938)
2 D W Carney et al, J. Am . Chem. Soc., 2014, 5, 1922 (DOI: 10.1021/ja410385c)
3 J K Munk et al, Antimicrob. Agents Chemother., 2014, 58, 1063 (DOI: 10.1128/aac.02369-13)
4 N Srinivas et al, Science, 2010, 327, 1010 (DOI: 10.1126/science.1182749)







No comments yet