
For the first time, chemists have used quantum mechanical calculations to predict the best of several possible retrosynthesis routes. This computational trick has allowed them to shorten the total synthesis of a six-ring terpene from 27 steps to just nine.
Relying on literature precedent or human intuition when designing a total synthesis can be tricky. It is hard to predict how compounds with multiple functional groups or unusual steric properties react. Unlucky chemists might spend months putting together a natural product one reaction at a time only to discover that a single step at the end of a long sequence doesn’t work. This often means starting the entire synthesis from scratch.
In a preprint study published on ChemRxiv, Timothy Newhouse and his team from Yale University, US, show that density functional theory (DFT) modelling takes the guesswork out of total synthesis.
DFT calculations model interactions between atoms and estimate how different compounds behave during a reaction. But modelling is usually done in retrospect, for example to study reaction mechanisms – Newhouse decided to use its predictive power.
Tricky terpenes
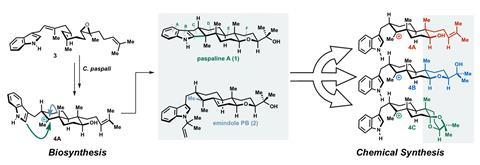
The team tackled two terpene syntheses: paspaline A and emindole PB. These fungal terpenes disrupt signalling pathways in certain cancer cells. The former’s synthesis had previously taken 27 steps – the team did it in nine – while the latter one had never been made.
Paspaline has a steroid-like structure with an indole fused to the cyclopentane ring. Previously, chemists built the indole rings over several steps right at the end of the total synthesis. Inspired by paspaline’s biosynthesis, Newhouse and colleagues decided to attach an off-the-shelf indole to the half-formed steroid frame. A tricky cyclisation at the very end of the synthesis would then complete the final steroid ring – a risky move, says PhD researcher and first author Daria Kim.
To choose the best out of three possible precursors for this crucial transformation, the team performed DFT calculation on how substituents at the molecule’s far end influence the reaction outcome. A compound with a bicyclic ketal substituent turned out to have the most favourable energetics – a somewhat surprising result given that it was more sterically strained than the other options.
‘[DFT calculations] allow us to evaluate substrates that are modified in ways that don’t obviously change the outcome of a transformation,’ explains Newhouse. ‘The functionality that we modify can be very distal from the reaction site and doesn’t seem like it would have such an impact on the course of a key step – but it does.’
DFT made another unusual prediction that turned out to be right: the compound would form a trans-fused ring system, although reactions reported on similar substrates had led to cis-fused rings.
Solvent in a haystack
However, DFT can’t predict which solvent would work best or at which temperature to run the reaction. ‘Each of the substrates we looked at is like a haystack, and in one haystack there is a needle [for the best reaction conditions],’ explains Kim. ‘The process of extracting that needle is still very difficult and time consuming, but with DFT you have more of an assurance that your haystack does contain that needle.’
Moreover, Kim explains, DFT only works if users know the reaction’s mechanism. ‘The calculations are only as good as the information you put in. DFT is not going to identify a mechanism for you.’
‘[This method] would allow for multiple hypotheses to be tested and to help prioritise which routes could be most viable,’ says postdoctoral researcher Carolyn Ladd, who works on natural product synthesis with Sarah Reisman at the California Institute of Technology, US. Since many chemists already use DFT software, it should be straightforward to adapt it to retrosynthesis planning, she adds. ‘It is a great way to take advantage of computational methods that have already been well-established.’
‘It’ll be interesting to see if computationally pre-screening potential substrates is something that will catch on in the wider community,’ says Kim. Newhouse thinks that DFT calculations could even increase the predictive power of synthesis planning tools such as Chematica.
References
D E Kim, J E Zweig and T R Newhouse, ChemRxiv, 2018, DOI: 10.26434/chemrxiv.7322330.v1














No comments yet