
Classic hydrogenation catalyst installs multiple stereocentres one by one to create small amino acid rings as single enantiomers
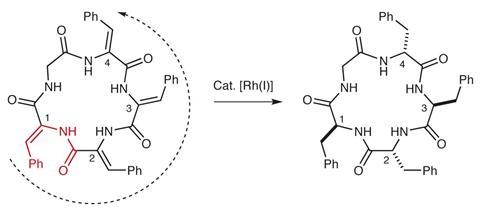
A rhodium catalyst that can walk around a macrocyclic ring and create multiple stereocentres in a row could change the way chemists make cyclic peptides. The unusual mechanism was discovered in a hydrogenation catalyst that has been studied for half a century.
Cyclic peptides can be designed to adopt particular shapes – a feature scientists are hoping to harness for designing new antibiotics and immunosuppressant drugs. To make peptide rings, the individual amino acids are first linked, then the chain ends are brought together. But this isn’t always straightforward.
‘The big problem with current methods is that the actual cyclisation of the peptide is very difficult – it’s very sequence specific, often low yielding and there are a number of side reactions, in particular epimerisation [stereochemistry scrambling] of the C-terminal amino acid,’ explains cyclic peptide expert Katrina Jolliffe from the University of Sydney, Australia, who wasn’t involved in the work. Stereochemistry dictates biological activity, so chemists can’t allow any reactions messing with the atoms’ spatial arrangement.
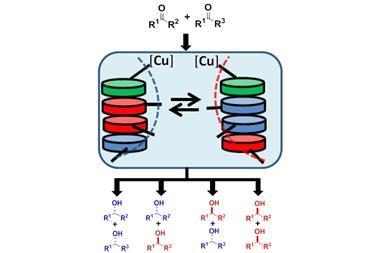
Vy Dong and her team from the University of California Irvine, US, have come up with an idea that avoids this problem: starting with entirely achiral building blocks, they use a hydrogenation catalyst to install every single stereocentre after cyclisation.
Although the rhodium catalyst itself isn’t chiral, hydrogenating a four-residue ring produces only one pair of enantiomers rather than a random mix of all 16 possible stereoisomers. To track down the origins of this curious stereocontrol, Dong worked together with mechanistic chemist Olaf Wiest, from the University of Notre Dame, US.
It turns out that the catalyst creates the stereocentres sequentially, one after the other, with the previous stereocentre controlling the arrangement of the next one. ‘[The catalyst] knows exactly where to go based on the flexibility of the residue one before [the one the catalyst is currently reducing],’ explains Wiest. Adding a chiral ligand makes the reaction entirely enantioselective.
‘I think it’s a powerful approach to complement what is currently used in peptide synthesis,’ says Andrei Yudin, an organic chemist at University of Toronto in Canada. ‘But we have to remember that making peptides from amino acids via amide coupling is such a mature technology, the barrier is very high to disrupt it.’
‘What we found is the main bottleneck, surprisingly, is not the hydrogenation but the pre-cyclisation strategy,’ says Dong. However, carrying out the dehydropeptide synthesis on a solid support like regular peptide synthesis, which Jolliffe thinks should be possible, might make it easier for chemists to adopt the method.
The catalyst’s surprising behaviour, Dong and Wiest point out, goes to show that there are still new things to discover about well-known reactions. ‘Hydrogenation is such a classic reaction, it’s neat to see an old catalyst doing something very new, being involved in a sort of molecular recognition process, communicating with the substrate,’ says Dong.
References
D N Le et al, Nat. Chem., 2018, DOI: 10.1038/s41557-018-0089-5










No comments yet