Flexibility and creativity help overcome unexpected obstacles
Nature produces an incredible variety of cyclic compounds, from tiny cyclopropanes and epoxides, to giant macrocycles of 50 atoms or more. However, the more familiar five- and six-membered rings remain by far the most common in natural products. That’s not too surprising, since these occupy a special ‘Goldilocks’ space, much less strained than their larger or smaller cousins. This makes them much easier to synthesise (whether by a chemist or an enzyme).
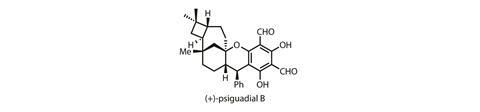
Four-membered rings are particularly uncommon, as nature has relatively few good options for putting these spring-loaded cycles together – most are biosynthesised using cationic reactions or cycloadditions. Unfortunately, things aren’t much easier for the synthetic chemist. In the laboratory, cycloadditions are by far the safest option, but it’s not always easy to get the required substitution, stereochemistry or regioselectivity. If a cycloaddition won’t do, chemists must get creative, as Sarah Reisman and co-workers at the California Institute of Technology, US, have done in their synthesis of the unusual natural product psiguadial B.1
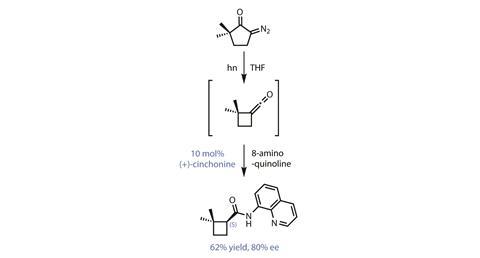
The group’s bold strategy is to create the cyclobutane first, employing a Wolff ring contraction of a diazocyclopentanone (figure 1). Coupling this with some clever asymmetric organocatalysis delivers the chiral cyclobutane building block with good selectivity. However, this impressive tandem reaction actually gives the wrong enantiomer. Unfortunately, by far the best organocatalyst for the asymmetric ketene addition is the alkaloid cinchonine. Only one enantiomer of cinchonine occurs naturally, so the team cannot simply use the oppositely configured catalyst to reverse the selectivity.
However, one reason that cinchonine is a popular choice for catalyst development is that the isomeric natural product cinchonidine tends to behave as if it were the enantiomer of cinchonine (it isn’t quite, so it’s called a pseudoenantiomer). Sadly, in this case, cinchonidine reacts quite differently, with much lower selectivity.
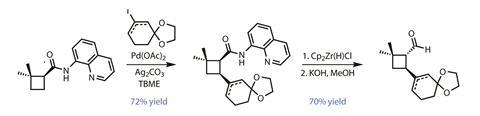
Rather than re-engineer the route to deliver the correct stereochemistry, the team changed tack and developed a plan to flip it to the correct configuration later in the synthesis. The group used the newly installed aminoquinoline amide to direct a palladium-catalysed C–H vinylation (figure 2), adding a protected cyclohexenone group onto the cyclobutane ring, cis to the amide. Reducing the amide to an aldehyde and adding base allows the substituents to flip to the more stable trans-configuration, neatly inverting the problematic stereoselectivity from earlier.
From here, seven more steps complete the pentacyclic core structure of the natural product, and the team is left with just the phenyl and aldehyde groups to install. Again, they elect to use a C–H functionalisation to speed things up. First, oxidising the extremely electron-rich benzylic carbon installs a temporary ether group (figure 3), which is then displaced with a phenyl cuprate to attach the final ring. Finally, cleaving the methyl ethers and a double formylation complete the target.
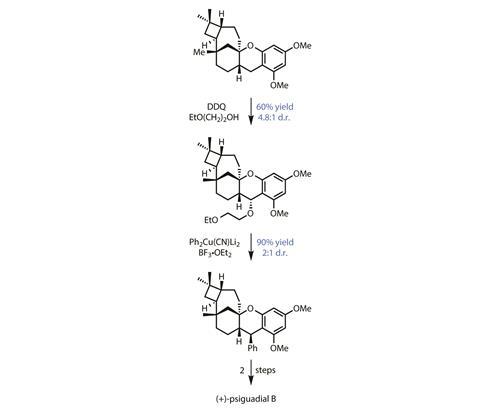
Thanks to the group’s decision to tackle the challenging trans-cyclobutane ring system at the start of the synthesis, and judicious exploitation of innate C–H reactivity to speed up carbon–carbon bond forming steps, this tricky target is completed in just 15 steps overall.
References
L M Chapman et al, J. Am. Chem. Soc., 2016, 138, 9803 (DOI: 10.1021/jacs.6b07229)











1 Reader's comment