
Phil Baran is spurring organic chemists to rethink how they make complex compounds, as Mark Peplow discovers
‘Nothing really inspired me until chemistry came along – it was love at first sight.’ This love affair has consumed US chemist Phil Baran of the Scripps Research Institute in La Jolla, California for more than 20 years, and propelled him to the forefront of organic chemistry.
At the age of just 36, he has already racked up some of the most lauded natural product syntheses in history, and won dozens of the field’s highest accolades. Last year, he was awarded a MacArthur Fellowship – often referred to as a ‘genius grant’ – worth $625,000 over five years. ‘He’s the best synthetic chemist of his generation,’ says David Schuster of New York University in the US. ‘And he’s a helluva nice guy.’
Perhaps his greatest contribution, though, is to shatter the myth that making large, complicated molecules is a purely academic exercise in grinding out just enough product to characterize. Above all, Baran is a practical man: his syntheses tend to be extremely efficient, and are designed with scale-up in mind. ‘The ultimate expression of a natural product synthesis is to make it in gram quantities, and invent methods that have wider uses,’ he says.
Chemistry addict
Baran was born in Denville, New Jersey, on 10 August 1977, and grew up in Coral Springs, Florida, where his family moved to help ease his brothers’ asthma. He remembers being a poor student at high school, preferring to play role-playing games, write computer programs and build with Lego. The common theme was ‘creating what didn’t exist before’, he says.
Encouraged by his chemistry teacher to experiment after school, Baran quickly channeled his creativity into crafting molecules. In 1995 he began a chemistry degree at New York University, and enthusiastically accepted Schuster’s offer to work in his lab, synthesizing compounds that linked C60 with porphyrins to make artificial photosynthetic systems.
Learning from EJ Corey is like a Jedi warrior learning from Obi Wan
Phil Baran
‘He was here all the time,’ recalls Schuster, who would sometimes find that Baran had spent the whole night in the lab. ‘I’d come in in the morning, and on my desk would be a vial, or an NMR spectrum, of something he’d just made. He was an incredible student.’
Baran parent’s divorced when he was young, and his all-consuming interest in chemistry had left little space for wider interests. Schuster took him to performances of the New York Philharmonic, and introduced him to sport – realizing in the process that Baran’s extremely poor eyesight had never been addressed. ‘Dave took me under his wing and was like a father figure; he turned me into a presentable human being,’ says Baran.
After attending a lecture given by K C Nicolau, now based at Rice University in Houston, US, ‘Phil turned to me and said “That’s the guy I wanna work with”,’ says Schuster. The PhD at Scripps with Nicolau that followed was ‘like hardcore Navy Seal training’, laughs Baran. In 2001, he moved to Harvard University in Cambridge, US, for post-doctoral research with E J Corey. Corey had won the Nobel prize in chemistry in 1990 for his development of retrosynthetic analysis – the art of logically dissecting complex molecules on paper in order to better plan their construction in the lab. ‘Learning from him is like a Jedi warrior learning from Obi Wan,’ says Baran.
‘When he was in my labs he made a very profound impression on me, despite his young age. He had a phenomenal grasp of synthetic chemistry,’ says Corey. ‘I felt that he could be a leader in his generation.’
Guiding principles
Baran returned to Scripps as an assistant professor in 2003, and three years later won tenure at the remarkably young age of 28 years old. He’s been there ever since, building a rapidly-growing list of natural products. In 2006, for example, he created the potent anticancer agent haouamine A, the marine alkaloid chartelline C, and several more complex natural products.
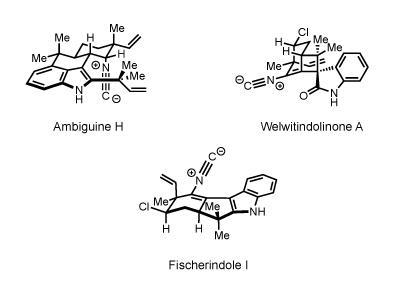
But Baran was just getting warmed up. The following year, he published a landmark paper that served as a manifesto for his approach to synthesis, and applied it to create a range of enantiomerically pure marine natural products, including ambiguine H, welwitindolinone A and fischerindole I, in gram quantities.
The impressive syntheses were underpinned by eight guiding principles. Baran eschewed protecting groups, for example, which are commonly used in organic synthesis to safeguard completed sections of the molecule while other parts are being built. Protecting groups are incredibly useful tools – but adding and removing them adds extra steps to a synthesis and can reduce yields dramatically. By exploiting the innate reactivity of functional groups in the molecule, his syntheses avoided protection altogether.
Baran also used as few redox reactions as possible, instead designing the syntheses so that the oxidation state of the molecule changed gradually and consistently from one intermediate to the next. The reactions were also chosen to maximize the number of carbon–carbon bonds formed in each step, sometimes by exploiting cascade reactions that zipped up large sections of the molecule at one fell swoop. The result was a series of efficient syntheses that could make plenty of the compounds for biological testing, and in a way that allowed ready access to analogues with the same carbon skeleton. ‘It’s not just about grinding something out in 30 steps – he’s interested in a more elegant approach,’ says Schuster. Although other chemists had used each of these principles separately for years, his rigorous adherence to all of them within the same syntheses raised the bar for total synthesis, Schuster adds. ‘He just does it better than anyone else.’
Total synthesis papers often talk up the bioactivity of the molecules and their potential drugability, yet all too often the final route will only ever be suitable for producing milligrams of material, with no easy way to produce analogues. Baran says his broader goal was to prove that natural products could be made in practical quantities for routine use in drug discovery.
The ‘eight commandments’ drew some flak at the time, says Baran, not least from a few senior chemists who had devoted their life’s work to developing protecting group strategies. Some pointed out that he has chosen targets that are particularly well suited to minimizing protecting group chemistry, and that producing molecules like polysaccharides will always need protection strategies.
But Baran notes that in the seven years since that paper appeared, there have been more than 100 total syntheses that are proudly protecting-group free, along with an increased focus on efficient synthesis (see Stepping towards ideality). ‘It’s a sea change in the community as a whole,’ he says. ‘Efficiency is the future’
The industrious chemist
In 2009, Baran cracked one of the toughest problems in natural product synthesis. The contorted alkaloid palau’amine, originally discovered in the Pacific Ocean sponge Stylotella agminata, bears nine nitrogen atoms that are just aching to react in disruptive ways, and eight sterogenic centres whose bonds must be positioned just so. One wrong move, and the whole strained structure springs open like a trap.
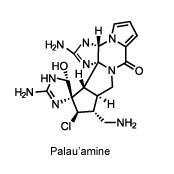
For more than a decade, palau’amine remained unconquered as organic chemists tried and failed to find a route to build it in the lab. It didn’t help that they had been chasing an incorrect, bowl-shaped structure for years – Baran was part of an international team that pointed out that error in 2007.
His synthesis once again espoused the principles of redox economy, and featured trademarks such as a five-reaction cascade and no protecting groups. The synthesis also introduced a novel, highly selective method for adding a hydroxyl group to a sensitive corner of the molecule, using silver(ii) picolinate. To round off the synthesis, Baran’s team created an unusual nine-membered ring that could be pinched together to form the fused ring system at the heart of the final product. An enantioselective synthesis soon followed. ‘The ability to design a synthesis like that is a sign of a mind that cuts like a knife,’ says Corey.
The total yield of 0.015% was arguably not much to write home about, but Baran says that the methods developed for the synthesis have found widespread use in drug development projects in the pharmaceutical industry. He should know – Baran reckons he spends about 10–15% of his time consulting for industry, which he says is ‘absolutely essential’ for his research. ‘If you don’t find out what they need, it’s a handicap.’ Ever the practical chemist, he is less interested in impressing colleagues in academia than he is in providing tools for the far larger numbers of chemists working in the pharmaceutical and agrochemical industries.
In 2011, he co-founded Sirenas Marine Discovery in San Diego, US, to generate drug candidates inspired by marine natural products. And last year he released an iPad book, The portable chemist’s consultant, co-authored with his wife Ana Montero and lab colleague Yoshihiro Ishihara (reviewed in Chemistry World). So far, they’ve sold more than 500 copies, mostly to people working in companies in more than 20 countries worldwide.
Although his total syntheses get the most press, Baran has also developed a startling array of new reactions that are used in industry. In 2011, for example, he showed that sodium trifluoromethanesulfinate (CF3SO2Na), a stable, inexpensive solid, could add a trifluoromethyl group to an aromatic ring. Trifluoromethyl groups are often used to fine-tune a drug’s interaction with proteins or improve its biological stability, but traditional methods to introduce them had relied on difficult-to-handle reagents such as anhydrous hydrogen fluoride or gaseous trifluoromethyl iodide. An equally simple method for adding difluoromethyl groups, using zinc dialkylsulfinate salt (Zn(SO2CF2H)2), followed in 2012. Sigma-Aldrich has commercialized the reagents, and Baran says that they are selling well. ‘I can’t think of anyone who has invented more broadly in synthetic chemistry,’ says Corey.
Taking on taxol
It was an industry partnership that led Baran to his latest high-profile synthesis. Ingenol is a terpene found in the milkweed plant Euphorbia peplus that has anticancer activity in animal models. In 2012, the US Food and Drug Administration approved ingenol mebutate, marketed as Picato gel, to treat actinic keratosis, a skin condition that can lead to skin cancer.
Ingenol mebutate can be extracted directly from E. peplus, but a kilogram of plant matter delivers just 1.1mg of the compound. A kilogram of dried seeds from another plant, E. lathyris, can deliver 275mg of ingenol itself, but this is still an extremely inefficient process. Baran’s group had already refined a general strategy for terpene synthesis, and so Picato’s manufacturers, Danish company LEO Pharma, asked him to come up with a total synthesis of ingenol that was short enough to scale up commercially, and that could also deliver analogues.
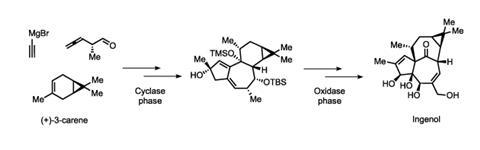
Two previous ingenol syntheses had required 37 and 45 steps, but Baran’s route whittled that down to a mere 14, conceptually divided into two phases. In the first ‘cylase’ phase, Baran’s team built up the core carbon skeleton of ingenol from the simple commodity chemical (+)-3-carene, producing more than a gram of a key intermediate. In the second, ‘oxidase’ phase, they decorated the molecule with four side-by-side hydroxyl groups that had previously posed a major problem in other syntheses – as well as finishing off the molecule’s ring structure with what they described as ‘a seemingly improbable’ rearrangement reaction.
LEO Pharma has already scaled up the synthesis to make more than one kilogram of ingenol, and Baran hopes that it will soon replace the plant-derived production method. Ready access to that key intermediate has also enabled the company to develop ingenol derivatives that are more potent, potentially broadening the range of cancers that they might treat.

Dividing the route into these two phases mimics nature’s strategy of compartmentalizing the synthesis, says Baran: first build the carbon skeleton, which has few groups that need protection, and then functionalize it. He’s using the same approach to tackle one of the most iconic terpenes of all: the anticancer agent taxol. ‘Nature can make tonnes of taxol,’ says Baran, but 10 total syntheses have collectively managed a grand total of 28mg. His group is now preparing a paper outlining the first stages of a synthesis that should be able to produce synthetic taxol on a large scale. ‘We would like to make 1g of taxol in the lab in a procedure achievable by one student,’ says Baran.
‘Slow down, Phil!’
Baran still puts in long hours at the lab, arriving for work at 6am every morning. In 2011, a tongue-in-cheek letter appeared online imploring him to ease up. ‘On behalf of all mankind: Would you please slow down?’ it read. ‘We mortals have no chance to keep up with you. It is not funny anymore. We need a break.’
But the undergraduate all-nighters are in the past – these days, he’s home by 6.30pm to see his wife and three children. Still, Schuster frets a little over his welfare, like a concerned parent. ‘He travels too much. I said to him, “Turn some things down”.’ (That advice didn’t stop Baran giving three talks in one day at the spring meeting of the American Chemical Society earlier this year.)
In the future, ‘I can see him going into a department where he would have the job of building up a synthetic organic chemistry programme and hiring top people,’ says Schuster. ‘I don’t see him going into administration though, that would take him away from what he loves doing.’ Baran himself is adamant that his only goal is to continue producing useful, innovative chemistry.
That, at least, seems certain. ‘If you project out in the future,’ says Corey, ‘his lead over the competition will be eye-opening 20 years from now.’
Mark Peplow is a science journalist based in Cambridge, UK










No comments yet