
More oversight is needed over products in the grey area between medical device and medication
Powders, tablets and gels used like medications but registered as medical devices – the same category plasters and pacemakers fall into – need tighter controls, a team of clinical researchers has concluded after studying several such materials. Products that fall into this regulatory grey zone don’t need to prove their efficacy or safety in the same way pharmaceuticals do, which could be a concern for vulnerable patients like children.
‘Patients were telling me that they were taking medication – Ziverel – and I didn’t know what it was,’ recalls Myriam Van Winckel, a paediatric gastroenterologist at Ghent University Hospital, Belgium. ‘I didn’t find it in the list of medications that are available in Belgium. It was a medical device, not a medication, despite the fact that it was presented like one.’
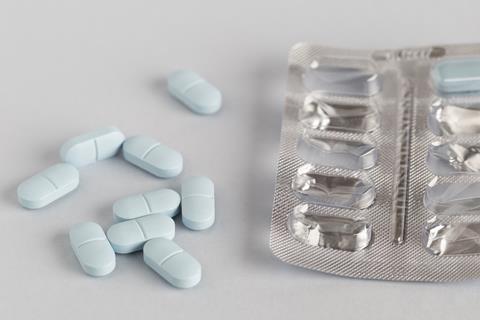
According to European regulations, medical devices are used for diagnosis, prevention or treatment of disease, but unlike drugs their primary mode of action isn’t biological. The definition includes everything from medical software to wound dressings and surgical instruments. But it also applies to products like Ziverel, which contains hyaluronic acid, the polysaccharide chondroitin sulfate and a polyoxypropylene–polyoxyethylene copolymer.
Like a drug, the acid reflux-relieving material is taken orally several times a day. The reason Ziverel and similar products aren’t classed as drugs is because the makers argue that its main mode of action is creating a mechanical barrier along the oesophagus, Van Winckel explains. ‘But they don’t have to prove that,’ she adds. ‘Indeed, trying to define what’s a barrier is very difficult.’
‘Ziverel has a mechanical mode of action forming a bioadhesive film over the gastro-oesophageal mucosa, thereby protecting it against the action of the hydrochloric acid produced by the stomach,’ a spokesperson confirmed in a combined statement from APharm and Norgine, the product’s manufacturer and distributor, respectively. ‘Product safety and efficacy has been demonstrated by different published clinical studies,’ the spokesperson adds.
They are most likely medicines, not medical devices, because the principal mode of action is pharmacological, not physical
Manufacturers need to demonstrate medical devices’ safety and performance. An organisation called a notified body then assesses the product’s conformity and grants it a CE mark. But Van Winckel and her colleagues found only poor clinical evidence for the three devices they investigated. Studies were small or of low quality, often missing placebo controls. In some cases, there was no data at all, for example to show the effect of Ziverel in children.
The researchers also highlighted the lack of safety information in the devices’ instruction leaflets. There was no or little mention of interactions with other compounds, maximum safe dose or adverse effects. ‘On some websites, for example for Ziverel, they mention that it’s not allowed for patients under the age of three, but this is not mentioned in the leaflet,’ Van Winckel says. ‘It has been promoted to paediatricians without mentioning that.’
While Van Winckel doesn’t know of any instances in which such medical devices have caused harm, but says that ‘it is difficult to be to be sure of that’.
However, Val Theisz, director of regulatory and clinical affairs at the Medical Technology Association of Australia, thinks the study should have focused on managing incorrect medical device classification. ‘The examples provided are most likely medicines, not medical devices, because the principal mode of action is pharmacological, not physical,’ she says.
The leaflet for Tasectan Duo, for example, mentions the product ‘is used to restore the physiological function of the intestinal wall’. The phrase ‘restoring physiological function’, Theisz points out, appears in the definition for drugs. Not only did the manufacturer wrongly apply for regulatory approval as a device, ‘it appears that the [responsible] notified body failed to recognise that the products are medicines’, Theisz says.
‘In my opinion, the actual problem was not identified adequately therefore the proposed solution is not appropriate,’ Theisz continues. ‘There is no need for medical device regulations that mimic medicines regulations, because they would be inappropriate for medical devices. What is needed is effective oversight of EU notified bodies and this is likely to be significantly improved under the new regulatory regime for medical devices.’
From May 2020, the new regulatory regime will require manufacturers to consult with the European Medicines Agency on their medical devices classification. Devices in the highest risk category, which includes substances and implants, will also need to go through clinical investigations.
‘The issues raised in the report will be addressed by the new regulations,’ a spokesperson of the UK’s Medicines and Healthcare products Regulatory Agency tells Chemistry World. ‘We believe that these changes will place an appropriate level of additional scrutiny on these products to ensure their safety and clinical effectiveness.’
But Van Winckel thinks further revisions are needed. ‘There is still no requirement of efficacy, performance being sufficient,’ she points out. ‘An additional problem remains that there is no control, nor approval procedure, foreseen for the leaflet.’
‘We propose that all products intended for repeated internal administration – oral, nasal or rectal – should follow proper registration as a medicine through the European Medicines Agency,’ Van Winckel says.
References
S Huijghebaert et al, Arch. Dis. Child., 2020, 105, 147 (DOI: 10.1136/archdischild-2018-316391)














No comments yet