
US chemists have developed an approach that could simplify the currently extremely difficult process of custom-designing proteins for a specific job. Nick Polizzi and Bill DeGrado from the University of California, San Francisco (UCSF) used thousands of protein structures in the Protein Data Bank (PDB) to define a new structural unit. Rather than worrying about the exact positioning of side-chains of each amino acid residue in a protein, they shift focus to the chemical groups each residue is in contact with. Using their method they designed two different proteins from scratch that recognise the blood thinner drug apixaban.
Until now, the best protein-design algorithms needed many follow-up experiments to optimise the protein sequence to bind a compound like a drug. By contrast the UCSF team computationally designed and made just six sequences to find their two successful designs. ‘It is now possible to design proteins from scratch that bind complex small molecules,’ DeGrado tells Chemistry World. ‘We hope others will join with us in using and improving on the methodology.’
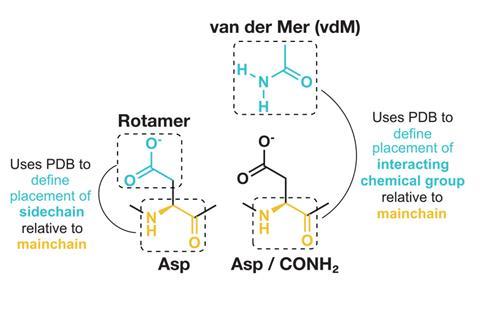
Polizzi and DeGrado’s approach uses a unit of protein structure called a van der Mer (vdM). The name combines two structural ideas. One is the van der Waals attractive forces that can hold together molecules in close contact. It also refers to rotamers, the different sidechain conformations amino acids adopt. A vdM maps each amino acid’s backbone to where nearby interacting chemical groups were found in the PDB. ‘Only now is the PDB large enough for us to define vdMs for each residue type and secondary structure,’ comments DeGrado. ‘It may sound very similar to earlier approaches, but the success of this work required a subtle shift of our frame of reference.’
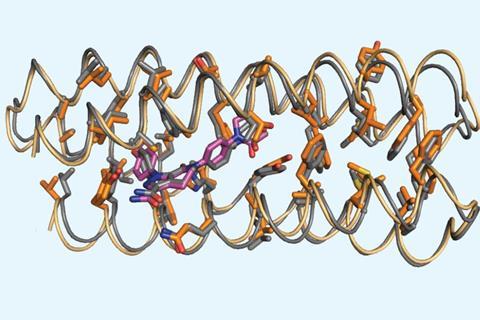
A UCSF cardiologist recommended targeting apixaban as a challenging binding target and because success could enable rapid reversal of its blood-thinning effects that might be needed in surgery. Two different protein bundles, one with a longer chain and different sequence than the other, both bound apixaban with dissociation constants in the low micromolar range.
Roberto Chica from the University of Ottawa, Canada, calls the results ‘very exciting’ and ‘at the forefront of the field of computational protein design’. ‘Importantly, this result was achieved without the need for downstream engineering of the designed proteins to enhance their stability or ligand-binding activity, in contrast with previous examples of de novo design of ligand-binding proteins by other groups,’ he says.
Polizzi notes that the design method could produce protein-based delivery vehicles and reversal agents for small molecule drugs. ‘We are also expanding the method to think about how we can design backbones as well as binding sites,’ he adds.
References
N F Polizzi and W F DeGrado, Science, 2020, DOI: 10.1126/science.abb8330














No comments yet