


Michael addition is one of the most well known reactions in organic synthesis. It is an immeasurably useful carbon–carbon bond-forming reaction involving the addition of a carbon nucleophile to an unsaturated carbonyl compound. Today, Michael additions are commonplace and Michael donors, acceptors and adducts are part of our shared vocabulary. Yet the story of its discovery deserves to be equally well known, because it marks a moment when mechanism first mattered to chemists.

Arthur Michael was born into a wealthy family in Buffalo, US, in 1853. His school did not teach chemistry, but he received private tuition and was provided with a laboratory at home in which to conduct his earliest experiments. On a trip to the newly unified Germany in 1871, Michael secured a position under August Hofmann at the University of Berlin despite having no formal qualification in chemistry. He spent the next nine years learning contemporary organic chemistry in the major laboratories of Europe, working with the likes of Robert Bunsen, Charles Adolphe Wurtz and Dmitri Mendeleev – as well as with Hofmann himself.1
Armed with a decent track record of publications and a passion for applying the principles of physical chemistry to understand organic reactions, Michael returned to the US. Settling in Boston, he began a career in chemistry that would be characterised by passionate self-determination. Despite holding no degree, he obtained tenure as professor of chemistry at Tufts College, and throughout his life he undertook long periods of self-funded research in his own laboratories. His was often a voice of criticism, railing against any new theory he considered to be unsubstantiated.
It was during his early period at Tufts that a report from Germany caught his critical eye: the reaction of sodium diethyl malonate with ethyl 2,3-dibromopropanoate (figure 1).2 The authors, Max Conrad and Max Guthzeit, proposed a reaction that we would understand today as a double substitution: the malonate species substitutes for both of the bromines in dibromopropanoate, resulting in a product that contains a cyclopropane ring. Cyclopropane itself was a very new molecule in the 1880s however, and Michael was sceptical.
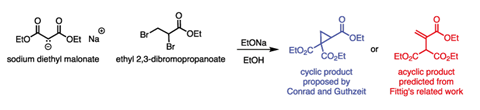
Structural assignment was a tricky business back then – the only tools available were elemental analysis, comparison with known compounds and referring to precedent reactions in the literature. Conrad and Guthzeit’s cyclic suggestion was based on an earlier report by William Perkin on the reaction of the same malonate salt with 1,2-dibromoethane.3 However, Rudolf Fittig had also reported that reaction and in his case proposed an acyclic, unsaturated product. The stage was set for someone to clear up the confusion.
Michael recognised that rather than having the reaction start with a substitution step, the first step could involve losing one of the bromine atoms as HBr. This would give the unsaturated molecule ethyl 2-bromoacrylate. Malonate would then attack – substituting for the remaining bromine – resulting in the acyclic (rather than the cyclic) product. To prove his theory, Michael took the second step of this process as his starting point – reacting sodium diethyl malonate with ethyl 2-bromoacrylate.4 When the product he obtained was the same as described by Conrad and Guthzeit, the Germans’ assignment of a cyclic product was disproven. Or was it?
Michael realised his experiment was flawed: he had assumed the malonate would substitute directly at the location of the bromine in the double bond. He now considered that it could attack at either end of the double bond (α or β attack), and that each route could give a different product. Thirty-five years before anyone thought to draw a curly arrow in anger, he worked through the possible intermediates and expected outcomes from these two reactions (figure 2) and saw that both the cyclic and unsaturated products were still possible.
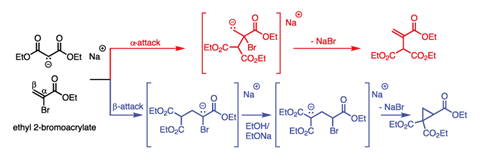
Drawing on the literature, Michael knew the reaction of hydrogen halides with unsaturated carbonyl compounds favoured β-attack. He therefore proposed that this reaction should proceed in the same way, and so lead to the cyclic product after all. He refers to the attacking malonate as ‘the negative part’ and the β-position of the double bond as the ‘relatively positive’ site, anticipating our modern electronic model of conjugate addition. As a definitive proof, he designed a synthesis of the known compound 3-phenylglutaric acid, which would only work if his theory was correct. It was.
So was born the Michael addition. But not everyone felt it deserved the name. Writing from Munich, Germany,5 Ludwig Claisen pointed out (robustly) that a precedent existed, in his own work with a young colleague named Telemachos Komnenos who had reported the β-addition of malonic acid or diethyl malonate to 2-butenoic acid four years earlier (as a side reaction in the synthesis of butenoic acid).6 Michael responded, defending his paper on its originality of thought; Claisen may have reported the reaction, but it was Michael’s meticulous investigation that proved how the reaction worked. Organic chemistry had one of its very first mechanistic studies.7
Soon after, Michael interrupted his work at Tufts with travel. As an art collector and lifelong enthusiast of ancient and medieval oriental cultures, he spent 18 months in Asia, Africa and Europe. The trip culminated in four years of unaffiliated research on the Isle of Wight in the UK, where Michael published widely to establish the scope of his reaction.8 Perhaps he did so with a note of defiance. In any case, his name remains.
References
1 E W Forbes, L F Fieser and A B Lamb, Harvard Univ. Gazette, 1943, 38, 246
2 M Conrad and M Guthzeit, Ber. Dtsch. Chem. Ges., 1884, 17, 1185 (DOI: 10.1002/cber.188401701314)
3 W H Perkin Jr., Ber. Dtsch. Chem. Ges., 1884, 17, 54 (DOI: 10.1002/cber.18840170111)
4 A Michael, J. Prakt. Chem., 1887, 35, 349 (DOI: 10.1002/prac.18870350136)
5 L Claisen, J. Prakt. Chem., 1887, 35, 413 (DOI: 10.1002/prac.18870350144)
6 T Komnenos, Justus Liebigs Ann. Chem., 1883, 218, 145 (DOI: 10.1002/jlac.18832180204)
7 A Michael, J. Prakt. Chem., 1887, 36, 113 (DOI: 10.1002/prac.18870360111)
8 T Tokoroyama, Eur. J. Org. Chem., 2010, 10, 2009 (DOI: 10.1002/ejoc.200901130)












No comments yet