
For the first time, chemists have used mechanical force to access unconventional trajectories on a reaction’s potential energy surface, making products that differ from the ones predicted by statistical thermodynamics. The researchers hope the technique can be exploited to discover new reaction schemes as well as to design probes of reaction dynamics.
Mechanical force can reshape the potential energy surface of the reaction, altering how thermodynamically favourable competing reaction pathways are, which in turn changes the product distribution. Chemists in the US have now performed ring-opening reactions of cyclobutane derivatives attached to long polymer chains either side. When they applied a shear force using the ultrasound, the polymer chains’ molecular masses dramatically affected the product isomer ratio. With a higher molecular mass, more alkene products were formed whose geometry mirrors the stereochemistry of the substituents in the starting cyclobutane ring despite the substituents being free to rotate – which should lead to Z and E alkenes being formed with equal likelihood.
‘The most obvious reason is because the potential energy surface was modified by force,’ explains Jeffrey Moore from the University of Illinois at Urbana-Champaign who co-led the study together with Stanford University’s Todd Martinez. ‘As the polymer chain gets longer and longer, the force that’s applied to the reacting molecule is greater and greater.’
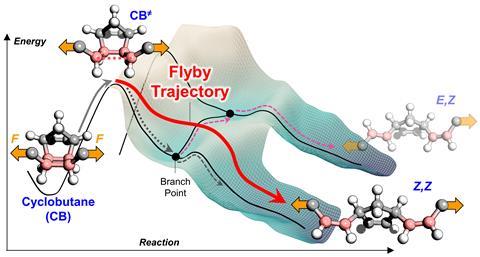
Martinez found their thermodynamic calculations did not match the experimental results. ‘It’s true that the potential energy surface changes under force, and that can have an effect, but that’s not enough to explain the isomer distribution shown here.’ Postdoctoral researcher Yun Liu – who works with Moore – suggested that, under force, the reaction might pass through a high-energy state of the intermediate directly from initial to final state without exploring all possible exits from the intermediate. This violates a central dogma of statistical thermodynamics that, as a reaction explores all paths, a lower energy path is always more probable. The researchers called these dynamic, statistics-disobeying paths ‘flyby trajectories’, which describes how the trajectories move over the energy barriers from an even higher energy state.
When the researchers altered the cyclobutane’s functional groups from alkyl to carbonyl groups to increase the intermediate’s stability, they saw ‘less product selectivity as the systems become more thermalised, because the barriers are now higher, blocking those flyby trajectories’, explains Liu. Dynamic effects have been identified in mechanochemistry and other organic reactions before, Liu says, but ‘the novelty here is that we are in control’. By first quantifying and then modifying how dynamic a system is ‘that really turns a notion into a handle that chemists can use to control reactions’, he says.
Mechanochemist Guillaume De Bo of the University of Manchester, UK, describes the results as ‘quite remarkable’. ‘Mechanochemistry allows you to reveal unexpected reaction pathways, either by modifying the potential energy surface, … or this one, which is really new, in which you play with how atoms move within the molecule itself to lead to a specific product,’ he says. ‘I suspect this is something that will be increasingly looked at, and that might be more general the more we look at it.’













No comments yet