
Nickel-guided radicals walk along carbon chains to make E-alkenes from terminal alkenes
A nickel catalyst takes radicals for a walk along carbon chains to transform terminal alkenes to E -alkenes. The reaction is so simple and robust that it might complement classic olefin metathesis – which produces Z -alkenes – in industrial applications.
‘The control of the stereochemistry of a carbon–carbon double bond is of fundamental importance in organic synthesis and numerous efforts have been dedicated to this endeavour over the last few decades,’ says organic chemist Ilan Marek from the Israel Institute of Technology, who wasn’t involved in the work.
Olefin metathesis – a reaction that redistributes double bonds between two molecules – was discovered in the 1970s and won a Nobel prize in 2005. It’s extremely popular both in industry, where it produces kilotonnes of olefins like propene or neohexene every year. However, it creates mixtures of olefin isomers that are difficult to separate from one another, although newer versions of the process can selectively access Z -alkenes.
There are a few ways to make E -alkenes, but most are far from general: some require a hydrogen atmosphere, others an expensive catalyst and most are restricted to molecules with few functional groups. ‘Although it might look like an old problem, new, efficient and reliable methods to fully control the selectivity remain an important issue,’ says Marek.
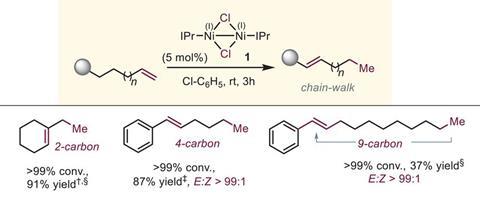
Franziska Schoenebeck’s team from RWTH Aachen University in Germany has created a nickel-catalysed reaction that relocates terminal double bonds to make E -alkenes.
In only three hours at room temperature, allylbenzenes are isomerised to the corresponding benzylic alkenes. The reaction converts eugenol into isoeugenol and estragole into anethole in more than 90% yield and with an E/Z ratio of 98:2. It can even transfer double bonds from outside to inside carbocyclic rings, like making methylcyclohexene out of methylenecyclohexane.
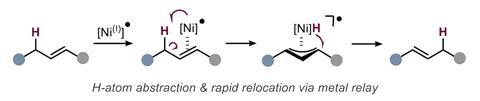
Responsible for the reaction’s stereoselectivity is a nickel(I) catalyst. In a single electron transfer, the catalyst grabs an allylic hydrogen atom. Nickel stays closely associated to the substrate in a metalloradical complex and relocates the hydrogen to form the energetically favoured E -alkene.
This mechanism, says Marek, is fundamentally different to other alkene relocation reactions. ‘Traditional approaches rely on metal–hydride addition and usually the final E/Z selectivity is not very high.’
In most molecules, the catalyst moves the double bond by one carbon atom. But if the substrate allows, it can also walk double bonds by two, three and four places. Even chain walks up to nine carbon atoms are possible – from 10-undecenylbenzene to E-1-undecenylbenzene – albeit with poor yield as other alkene isomers are formed as byproducts.
The reaction, says Marek, might be attractive to industrial chemists as it ‘combines operational simplicity, ease of purification, sustainability, scalability with functional group tolerance, short reaction times and mild conditions’. However, the catalyst is air sensitive and needs to be handled in a glovebox – a concern Marek hopes future research will address.
References
A Kapat et al,Science, 2019, 363, 391 (DOI: 10.1126/science.aav1610)











No comments yet