Scientists are decoding the brain’s exquisite molecular machinery – but there is still a long way to go
The slowly mounting agony of a black widow spider bite shows the fearsome damage that brain chemistry can cause. The arachnid’s venom hijacks the biochemical machinery that directs activity at the synapse junctions between neuron nerve cells in our brains. Like so much that happens in this system, we can partially explain the spider’s superpower, but still have much to learn.
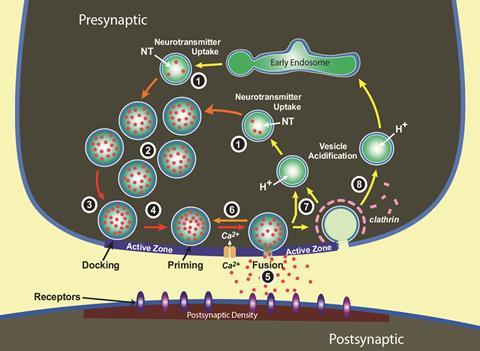
That’s because the mind-making molecules in our synapses do mind-blowing things. They transmit messages with incredible speed, enabling the wonders of human motion and reactions. When an electrical nerve signal reaches it, the synapse opens a channel that lets calcium ions in, mobilising spheres tens to hundreds of nanometres in diameter, known as vesicles. They then fuse with the fatty membrane around the synapse’s ‘send’ side, the pre-synaptic terminal.
After a delay that could be less than 100 microseconds, small vesicles release the neurotransmitters they encapsulate – a process known as exocytosis – in less than a millisecond. The neurotransmitters reach the post-synaptic terminal, directly opposite their release sites, triggering a signal to pass down the next neuron within a few milliseconds.
Nobody knew how this happened so fast when Thomas Südhof, now at Stanford University in the US, first started his laboratory in 1986. As he explained in the speech to accept the 2013 Nobel prize for physiology or medicine, he was determined to find out. ‘I was mesmerized by the apparent incomprehensibility of the speed of [calcium ion]-triggered release,’ he said.
Despite the impressive discoveries that followed, our ignorance still exceeds our knowledge, Südhof tells Chemistry World. ‘We don’t really know which of the many molecules that have been suggested in all these papers are actually involved’ in many synaptic pathways, he says.
Yet by teasing apart molecular roles, Südhof and other scientists have shed some light on our brains’ functions, such as memory, and its disorders, such as autism and Alzheimers’ disease. Today, there is an epilepsy medicine on the market that targets vesicle proteins. And black widow spider venom, along with similarly devastating toxins like those responsible for tetanus and botulism, has also offered several important clues.
Orchestrating a wild dance
Synaptic transmission is fast and precise thanks to the ingenious organisation of the ‘active zone’, where vesicles fuse with the pre-synaptic membrane. Active zone machinery docks and primes vesicles, bringing them near to calcium channels, poised to release neurotransmitters. After unleashing them in exocytosis, the machinery begins endocytosis, reconstructing and refilling vesicles ready for the next signal. Protein molecules enabling these processes are around a quarter of a vesicle’s spherical coat, with the remainder formed from fatty molecules like cholesterol.
Tetanus and botulinum toxins block neurotransmitter release by stopping synaptic vesicles fusing with the pre-synapse’s fatty cell membrane. Starting in the 1990s, Südhof and co-workers showed that they chop up the synapse proteins SNAP25, synaptobrevin-2 and syntaxin-1.1 To work out their exact role, Südhof and his team purified, analysed and identified many more synapse proteins and the genes encoding them.
This became a worldwide hunt, involving scientists including Eckart Gundelfinger at the Leibniz Institute for Neurobiology in Magdeburg, Germany. Purifying synaptic proteins from rat brains, ‘we stumbled over two very large pre-synaptic proteins’, Gundelfinger recalls. They then created gold-labelled antibodies that bound to these proteins in synapses in rat brain sections, showing up as dark spots under an electron microscope. These proteins, called piccolo and bassoon because of their need to work in harmony like an orchestra, only showed up near the active zone.2 ‘They are very large scaffolds that integrate many signalling pathways and processes in the presynapse,’ Gundelfinger explains. For example, bassoon helps precisely position certain types of calcium channels.
Such protein machinery increases or reduces the ‘strength’ of a synapse, a process known as plasticity that is linked to memory and learning. By moving calcium channels, bassoon can contribute to regulating neurotransmitter release, Gundelfinger explains, and in turn synaptic strength. ‘Maybe even more fascinating is that bassoon and piccolo can also integrate transcription repressors that regulate gene expression in the very same neuron,’ he adds. ‘This is very exciting – bassoon contributes to both short-term and, in particular through signals to the nucleus, to long-term plasticity.’
Kiss and run controversy
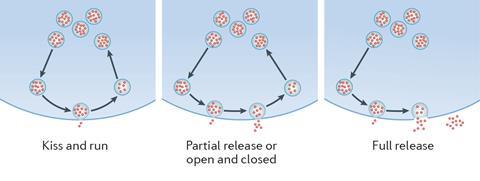
Today microscopy lets scientists like Ege Kavalali, from the University of Texas Southwestern Medical Center in the US, observe synapse proteins move on millisecond scales. By modifying them with green fluorescent protein, his team monitors neurotransmitter release during exocytosis and recycling in endocytosis. There are competing hypotheses about endocytosis, Kavalali explains. ‘One is that every time, you reproduce the vesicle from scratch.’ He calls assembling the various, very different, proteins a vesicle needs quickly enough ‘a daunting task’. The alternative is ‘kiss and run’, where vesicles fuse with the cell membrane and partially open, before closing again. While this is a controversial idea, Kavalali’s team has provided evidence of kiss and run-like mechanisms.3
More evidence comes from experiments that proved Andrew Ewing at Chalmers University in Gothenburg, Sweden, wrong. He set out to prove that vesicles always released their entire neurotransmitter load, while Gundelfinger, Südhof and others started hunting synaptic proteins. It took over a decade to develop the techniques needed, such as intracellular vesicle impact electrochemical cytometry (IVIEC). IVIEC pierces cell membranes with a 50–100nm diameter carbon fibre electrode, while voltages cycle from lower to higher values and back. This oxidises and reduces the chemicals released as the vesicles rupture, and the scientists measure the tiny resulting current changes, which indicate chemical concentrations.
Using IVIEC, Ewing’s team found that vesicles in isolated PC12 cells, a brain cell model from a rat tumour, contain around 114,500 neurotransmitter molecules, or 19 zeptomoles.4 But when the researchers triggered exocytosis, electrochemical measurements outside the cell found only around 73,200 molecules. Experiments like this show that most exocytosis events empty vesicles more thoroughly than usually expected in kiss-and-run – but not completely, Ewing says. He explains that the machinery Südhof revealed in his Nobel prize work influences how frequently the pores open to let vesicles release neurotransmitters. But how they close is more important for determining how much neurotransmitter they release, he adds.
‘Pore size and closing are clearly regulating these events,’ Ewing says over lunch at the Monitoring molecules in neuroscience conference in Oxford, UK, in March 2018. ‘I’m convinced that regulation involves the membrane changing as the vesicle is released. The vesicles have a different membrane composition than the plasma membrane around the cell. Some of that membrane is exchanged.’
Such membrane composition changes are a molecular memory that might influence plasticity, Ewing speculates. Membrane composition changes would also help explain how drugs and diet can influence synapse function, he adds. ‘If I eat this sandwich, it’s going to make a difference,’ he explains. For example, zinc is thought to regulate learning. Ewing’s team has found that vesicles in PC12 cells exposed to zinc contain less neurotransmitter than those that have not been, but still release the same amount in exocytosis.5 Drugs like cocaine and methylphenidate also change vesicle content and release in a similar fashion.
Hard targets
Südhof, however, is sceptical. ‘There’s no real evidence that kiss and run can ever be so fast for synapse vesicles not to release all their contents,’ he says. But he’s also now researching areas with limited evidence himself, such as how plasticity and synapse organisation become impaired during neuropsychiatric diseases. And this is where the key component of black widow spider venom, α-latrotoxin, has provided an important avenue of investigation.
α-Latrotoxin forms channels that let calcium ions through the synapse cell membrane when there’s no nerve signal. That activates the release machinery, causing a dangerous neurotransmitter flood. To do so, it binds to proteins near the synapse, located on the presynaptic terminal. Early in their protein hunt, Südhof and his team tracked down the genes responsible for producing these sites, and named their products neurexins I and II.
In subsequent years, the scientists found that neurexins are not just there to help out black widow toxins. They bind with neuroligins on post-synaptic terminals, helping align neurons precisely, with a synaptic cleft of around 20nm. But they’re also linked to brain disorders. ‘There are mutations in many genes that predispose people to schizophrenia,’ he says. ‘But the one gene with the most frequent association, and a single gene as opposed to multiple gene relations, is neurexin I.’ Neurexin I is also strongly associated with Tourette’s syndrome and autism, he adds.6
But Südhof’s current work on the neurexins is limited to understanding their essential functions, which ‘tells us very little’ about what might be happening in these disorders, he admits. ‘Eventually, I think the trans-synaptic cell adhesion machinery, the release machinery and the post-synaptic machinery must all be linked. But we don’t really know that connection.’
It’s easy to imagine that a lot of things can go wrong in this system
Today, proteomics is finding many other molecules that are potentially significant in brain disorders. For example, Gundelfinger and colleagues used high-resolution mass spectrometry to characterise the proteins in ground-up mouse brain cells. Combining their own results with other proteomic studies, they published the SynProt synapse protein catalogue in 2012, when it had over 2700 entries.7 More than 130 cause disorders when they are defective. ‘Of many synaptic proteins where the community has looked at the function, often they’re involved in synaptic failure and brain diseases,’ Gundelfinger says. Mutations in the bassoon gene, for example, have very recently been linked to childhood epilepsy and neurodegenerative disease.
This is evidence of what Kavalali calls the synapse’s ‘exquisite machinery’. ‘It’s easy to imagine that a lot of things can go wrong in this system,’ he says. Kavalali wants to target drugs at the synaptic vesicle machinery to help preserve it or compensate for its deficiencies, because it is almost ‘not tapped at all’, he adds.8 One reason is a lack of high-throughput assays for screening large potential drug molecule collections against synapse proteins. ‘There is a bottleneck there,’ Kavalali admits.
Another problem is that ‘some components are quite ubiquitous’, like the protein targets of botox, the toxin responsible for botulism. By destroying synapse proteins and preventing signals being received, it can permanently relax muscles, making it useful both cosmetically and medically. But botox must be injected extremely carefully, because if it reaches the brain that relaxation can be fatal, stopping breathing. Kavalali suggests other drugs could be injected at a specific site, similar to botox. Or, drug developers could target synaptic proteins that only get made in certain cells, he says.
But there is already one novel drug that targets the synapse’s protein machinery. Levetiracetam, a new generation anti-epileptic, specifically binds to a synaptic vesicle protein called SV2A, which is thought to regulate neurotransmitter release. ‘This drug is taken up through endocytosis and binds to this protein in the vesicle,’ explains Kavalali. ‘It has a strong impact on really intractable epilepsies. But what it does functionally, we don’t yet understand.’
Shades of grey matter
The synapse’s protein machinery is also implicated in the disease with arguably the greatest unmet need of all: Alzheimer’s. There are no ‘disease-modifying’ drugs yet available, highlights Tara Spires-Jones from the University of Edinburgh, UK. Researchers have trialled at least 190 drugs, largely focussing on reducing a protein called amyloid-beta building up in the brain, an obvious Alzheimer’s hallmark. There, amyloid-beta and another protein called tau destroy synapse machinery, and eventually the synapse-less neurons die. Such proteins progressively spread through synapses and neurons, which is what Spires-Jones studies. ‘This is really important in Alzheimer’s disease, because we know that the loss of synapses is the strongest pathological correlate of cognitive decline,’ she says. ‘We think it’s actually driving the disease.’
Spires-Jones and her colleagues have just published research showing tau accumulates on synaptic vesicles of human Alzheimer’s patients.9 Her collaborators, led by Patrik Verstreken at KU Leuven in Belgium, used fruit flies and mice to show that a relatively obscure synapse machinery protein called synaptogyrin-3 mediates tau-induced synaptic dysfunction.
Tau normally helps stabilise the microtubules that carry the ions whose movement produces electrical nerve signals, and also might protect hibernating squirrels from hypothermia. But knocking out tau genes in mice protects them against Alzheimer’s synapse destruction. ‘There have been several studies showing that tau is important in the post-synaptic density for amyloid beta-mediated synapse degeneration,’ Spires-Jones says. ‘It being there at all implies that there’s something functional about tau in the synapses.’
There are patient groups that are really waiting for progress
Stopping tau proteins from spreading and continuing to injure Alzheimer’s patients’ protein machinery might allow them to form new synapses, Spires-Jones hopes. She suggests using similar monoclonal antibody drugs to those that have failed so far with amyloid-beta to target tau, and giving them earlier in the disease process. ‘But we’re a long way from being able to effectively do this,’ Spires-Jones adds.
Considering the complexity in our heads makes it obvious why this is such a common refrain from scientists studying synapse proteins. Our brains contain around a hundred million neurons, each connected to thousands of others through around 100 trillion synapses. It’s hardly surprising, therefore, that thousands of different molecules control them. Südhof calls figuring out the important ones ‘an important initial goal, but by no means sufficient’. ‘We’re not going to be able to move forward drug development if we don’t actually understand the fundamental biology,’ he points out.
Yet recent meetings with epilepsy sufferers in Malta have underlined to Gundelfinger the importance of seeking that knowledge. ‘All of a sudden it makes sense of what we are doing,’ he stresses. ‘It’s not just basic research, it’s applicable. There are patient groups that are really waiting for progress in understanding these diseases.’
Andy Extance is a science writer based in Exeter, UK.
References
1 H McMahon et al, Nature, 1993, 364, 346 (DOI: 10.1038/364346a0)
2 E D Gundelfinger et al, Front. Synaptic Neurosci., 2016, 4 (DOI: 10.3389/fnsyn.2015.00019)
3 O A T Afuwape and E T Kavalali, Methods Molecular Biol., 2016, 1474, 187 (DOI: 10.1007/978-1-4939-6352-2_11)
4 X Li et al, Angew. Chem. Int. Ed., 2015, 54, 11978 (DOI: 10.1002/anie.201504839)
5 L Ren et al, Angew. Chem. Int. Ed., 2017, 56, 4970 (DOI: 10.1002/anie.201700095)
6 T C Südhof, Cell, 2017, 171, 745 (DOI: 10.1016/j.cell.2017.10.024)
7 R Pielot et al, Front. Synaptic Neurosci., 2012, 4 (DOI: 10.3389/fnsyn.2012.00001)
8 Y C Li and E T Kavalali, Pharmacol. Rev., 2017, 69, 141 (DOI: 10.1124/pr.116.013342)
9 J McInnes et al, Neuron, 2018, 97, 823, (DOI: 10.1016/j.neuron.2018.01.022)










No comments yet