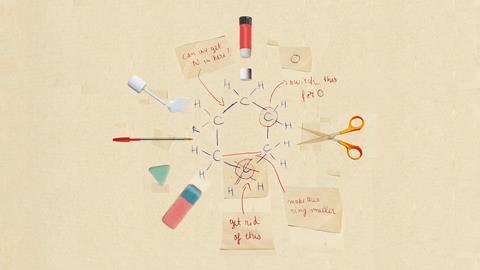
Nina Notman meets the chemists expanding the toolbox of reactions capable of adding, deleting and switching single atoms in rings at the heart of organic molecules
The majority of small molecule drugs have at least one ring system in their structural core. Sometimes the rings have biological activity themselves but more often than not their primary role is to offer structural rigidity. Rings have fewer degrees of freedom than linear molecules, meaning they can better hold their bioactive side chains in the orientation needed to fit in the active site of the target enzyme or protein. A 2014 analysis found that nearly 60% of small molecule drugs have nitrogen-containing heterocycles in their structure.
The drug discovery process typically begins with a high-throughput screen. A large library of molecules is tested against a biological target to see if one interacts with it in the desired way. Once a ‘hit’ is identified, medicinal chemists synthesise analogues of that molecule that are then tested against the target. The goal is to find a structure that is more selective, more potent or meets another requirement for the drug being developed.
The word editing evokes precision, the idea being that it’s not a drastic change
When shown the structure of a hit molecule, scientists can very rapidly draw up a wish list of analogues on paper that would be worth testing against the biological target. But whether medicinal chemists have the tools to make these molecules easily is a different matter.
Creating a new synthesis for each analogue from commercially available starting materials is both costly and time consuming. An alternative approach is to use the hit molecule as the starting point and then make controlled and precise edits to its structure. ‘The word editing evokes precision, the idea being that it’s not a drastic change to the molecule, it’s a really simple and precise reaction that allows you to make a very specific change,’ says Mark Levin, an organic chemist at the University of Chicago, US.
Changing things up
The vast majority of molecular editing reactions developed so far involve adding, removing or changing side chains on the periphery of the molecule. ‘This type of change happening on the periphery of the molecule is called a peripheral change or peripheral edit,’ explains Richmond Sarpong, an organic chemist at the University of California, Berkeley, US.
We want to think about inserting or deleting carbons, nitrogens and oxygens into a system of rings
Dozens of tools to perform precise and selective peripheral edits have been reported in recent decades, and more are being developed all the time. Examples include C–H functionalisation reactions, for converting inherently unreactive C–H bonds into C–C, C–O and C–N bonds, and cross-coupling reactions and click chemistry for joining fragments of molecules together.
An alternative – less used, so far – editing type involves making precise changes to single atoms in the structural core of molecules. This type of editing has been coined single-atom skeletal editing. ‘C–H functionalisation was a revolution that taught people to think about [editing] a part of the molecule that they were ignoring,’ Levin says. ‘What we’re trying to do is apply that same thought process to the heavy atoms in the molecule, so they can think about inserting or deleting carbons, nitrogens and oxygens into a system of rings.’
Changing the ring type in a molecule’s core can significantly impact its biological activity. When the number of atoms in a ring changes, so does the angle at which the side chains come off the ring. ‘This change in the way these groups are presented in space will more than likely also change the way [they] interact with the biological target,’ says Sarpong. Bioactivity can also be affected by switching the heteroatom without changing the size of the ring.
Sizing up
Sarpong and Levin are leading the effort to build a toolbox of general and precise skeletal editing reactions. The goal is to have a comprehensive collection of one-step reactions that can insert, delete and switch a number of heavy atoms (including carbon, nitrogen, oxygen, sulfur and phosphorus) in both aromatic and non-aromatic rings.
The toolbox already includes well-established ring expansion reactions including the Baeyer–Villiger oxidation, which inserts an oxygen atom next to a carbonyl group in a ring, and the Beckmann rearrangement, which does the same with a nitrogen atom.

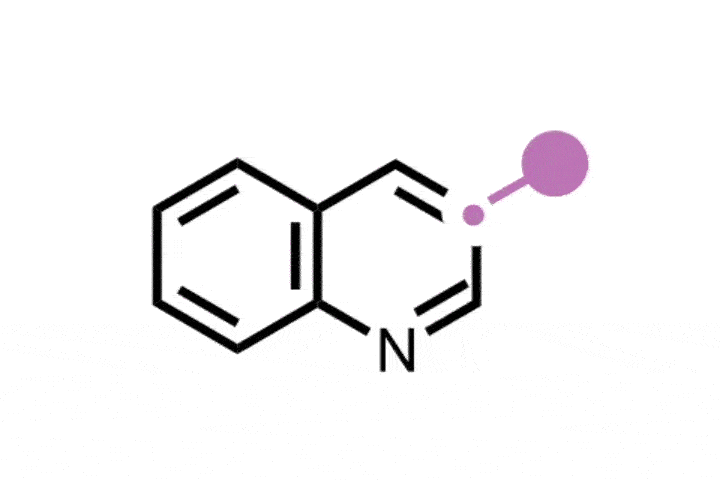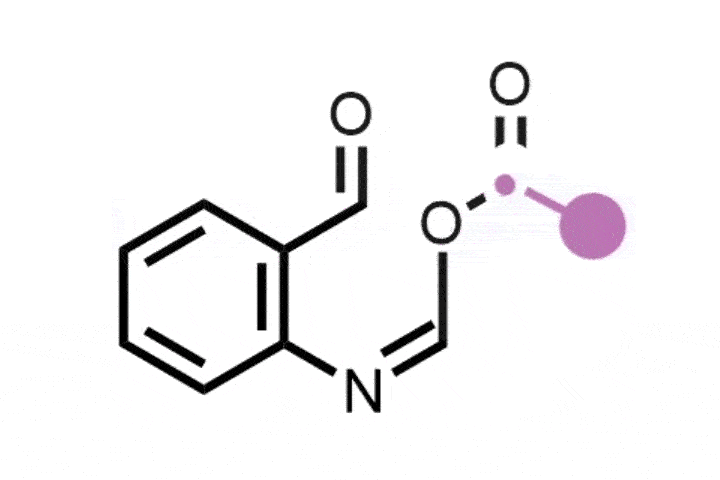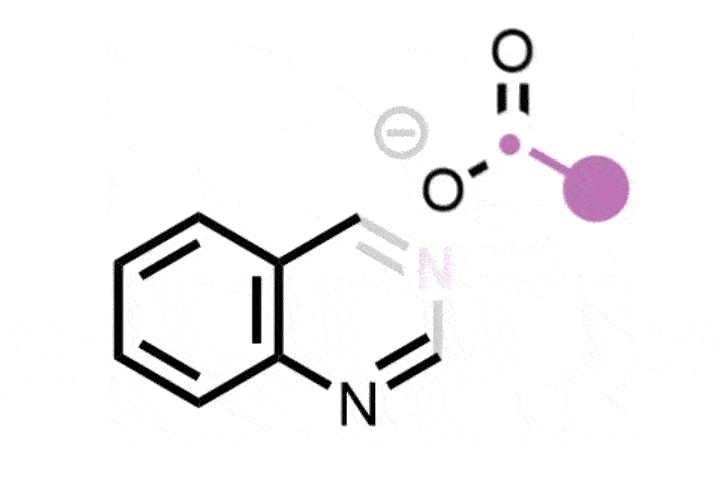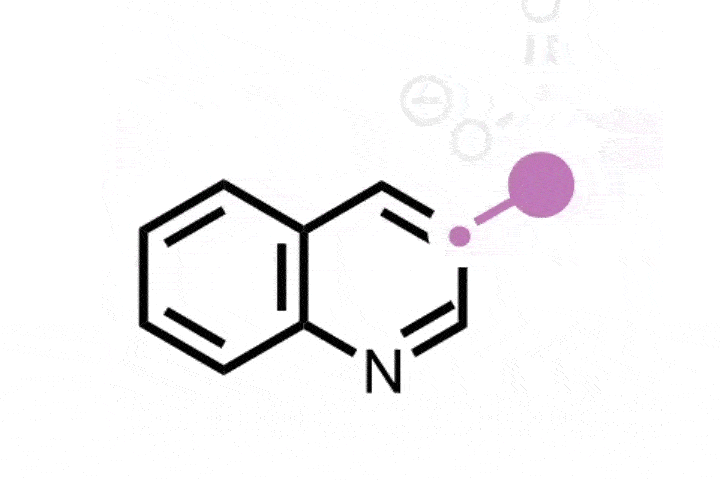
Newer tools include the Levin group’s 2021 protocol for inserting a carbon atom into five-membered pyrrole ring to make 3-arylpyridines, particularly important because pyridines are the second most prevalent nitrogen-containing heterocycle in drug molecules. Levin’s method is an improvement on the Ciamician–Dennstedt rearrangement. This reaction, first reported in 1881, inserts a chlorine-substituted carbon atom into a carbon–carbon bond in a pyrrole ring to make a chlorine-substituted pyridine. Levin’s approach uses chlorodiazirines as the chlorocarbene precursor to enable carbon atoms with a variety of aryl groups to be inserted, rather than just chlorine. He also used the same approach to transform indole scaffolds into quinolines.
In 2022, Julia Reisenbauer and Bill Morandi at ETH Zürich in Switzerland published a complement to the Ciamician–Dennstedt rearrangement, this time to insert a nitrogen atom into a carbon–carbon bond in the five-membered ring of an indole scaffold. This is achieved by generating an iodonitrene species in situ from a hypervalent iodine compound. The rearrangement tolerates a wide variety of functional groups as ring substituents and their position on the ring dictates where the new nitrogen atom is inserted, and therefore whether the end product is a quinazoline or quinoxaline scaffold.
In their paper, Reisenbauer and Morandi report nitrogen insertion into a wide range of indoles including the sleep aid melatonin and the beta-blocker pindolol. In a recent pre-print, the chemists reported using the same reaction to add a nitrogen atom into the five-membered ring of an indene. ‘It’s a different starting scaffold that we’re inserting the nitrogen into and then, of course, we are accessing a different class of compounds in the end,’ says Reisenbauer.
Sizing down
Tools are also being developed to make rings in the core of molecule’s one atom smaller. However, far fewer of these ring-contraction reactions have been developed so far than ring-expansion reactions. Contraction reactions fall into two categories: those that remove an atom from the molecule, and those that move an atom from the ring to a side chain on its periphery.
In 2018, Sarpong demonstrated the deletion of single carbon atoms from seven and six-membered saturated cyclic amines. In the former, an azepane ring turns into a piperidine, while in the latter a piperidine converts into a pyrrolidine. Piperidine and pyrrolidine rings ranked first and fifth, respectively, in the 2014 analysis of common drug structures.
We can extrude the nitrogen from the ring
The Sarpong group’s carbon deletion is a two-step, one-pot process. Dibromohydantoin and silver nitrate break the carbon–nitrogen bond in the cyclic amide and replace the carbon atom with bromine. Next, a strong base causes the carbon atom with bromine attached to join with the nitrogen and eject the bromine. The re-formed ring is one atom smaller than the original ring.
Sarpong’s group is also developing reactions that remove heteroatoms from saturated rings and into sidechains. In 2021, it reported, together with collaborators at Merck & Co, a light-mediated ring contraction reaction that relocates a nitrogen, oxygen or sulfur atom in a six-membered saturated ring to the molecule’s periphery. ‘We can extrude the nitrogen [or oxygen or sulfur] from the ring and contract it to make a five-membered ring,’ says Sarpong. The piperidine ring (in the case of nitrogen), tetrahydropyran ring (for oxygen) and thiane ring (for sulfur) are all transformed into a cyclopentane.
This two-step process requires an aromatic ketone side chain next to the heteroatom in the ring. First, the chemists use blue LED light to excite the ketone group, initiating a Norrish type II photochemical reaction that cleaves the carbon–heteroatom bond. The molecule then immediately closes up into a five-membered configuration via either a Mannich or aldol reaction when nitrogen or oxygen are removed respectively.
The chemists demonstrated this reaction on numerous molecules including on biologically active molecules with piperidine rings, including a cyclic derivative of the stimulant MDMC and a derivate of rimiterol, a bronchodilator.
Swapping success stories
To complement ring contractions and ring expansions, chemists are also starting to look at a third category of skeletal editing reactions – atom swapping. The ability to swap a single atom in an organic molecule was on the wish list of five reactions that organic chemists told Chemistry World they wanted in 2019.
An atom switch can occur in several different orders. In 2022, Junqi Li, an organic chemist at Iowa State University in the US, reported a method that inserts the desired atom into a ring before the unwanted atom is deleted. In this case, she was swapping a carbon atom with an oxygen atom to turn cyclic diarylmethane scaffolds into diarylethers.
Li’s reaction has three distinct steps. ‘We had an oxidation step [to oxidise a methylene group to a ketone], a Baeyer–Villiger reaction to insert an oxygen and then a decarbonylation reaction to remove the carbon atom,’ says Li. To achieve the third step, she needed to synthesise a novel nickel catalyst with a large bisphosphine ligand.
In 2022, Levin reported a way to approach the atom swapping challenge from the opposite direction – he removed the unwanted atom first before inserting the new one. In this case, he was replacing a carbon atom with a nitrogen atom. This reaction converts unsaturated quinoline scaffolds into indoles and then into the desired cinnoline scaffolds (with two neighbouring nitrogen atoms).
We accidentally found that this boron species can insert into THF
The carbon atom deletion process has multiple stages, with the key steps being a photolysis reaction that breaks the carbon–carbon bond followed by an acid-promoted rearrangement to form the indole scaffold. The insertion process is two steps: an amination reaction, to introduce an amine group, followed by an oxidative aromatisation to get to the cinnoline scaffold.
In 2021, Guangbin Dong, an organic chemist at the University of Chicago, US, took a different approach again – this time to switch an oxygen atom in a cyclic ether with a nitrogen to make a cyclic amine. This two-step process sees a boron atom inserted into the ether to make the ring larger by one atom, then the oxygen and boron atoms are replaced with a nitrogen atom.
Dong used dibromoboranes in the presence of a zinc/nickel catalyst to insert the boron next to the oxygen in the ether ring. This novel reaction was a serendipitous discovery, he explains. ‘We accidentally found that this boron species can insert into the solvent THF [tetrahydrofuran],’ Dong adds. The chemists went on to find that boron can be inserted into a wide range of five, six and seven-membered cyclic ethers, in the presence of various different substituents on the ring.
The switching of the boron and oxygen with nitrogen was achieved by an amination to insert the nitrogen into the carbon−boron bond followed by a dehydration to remove the unwanted atoms and reform the ring. Molecules Dong demonstrated his atom switch reaction on included the synthetic odour molecules galaxolide and ambroxide.
This nitrogen switch isn’t the only possibility once the boron is inserted into the ether ring. Dong also replaced the boron atom with a carbon to make a cyclic ether one carbon larger than it was to start with. His group demonstrated the utility of this reaction by synthesising the beta-blocker nebivolol in fewer steps than previously reported.
Editing polymer backbones
It was the invention of the Crispr-Cas9 gene editing system in 2013 that opened chemists’ eyes to the possibility of editing the skeleton of polymers – not only biological, but synthetic ones. As for small molecules, in the decades preceding this realisation a number of classical reactions like the Baeyer–Villiger oxidation had been applied to transform the skeletons of synthetic polymers.
‘The unification of these isolated examples into a field aimed to advance our ability to edit macromolecular skeletons is a recent phenomenon that is identical in spirit and synergetic with the developments in the small molecule chemistry,’ explains Alex Zhukhovitskiy, a chemist at University of North Carolina Chapel Hill, UK.
It would be great if we could take polyethylene and insert heteroatoms into its backbone
Skeletal editing on polymers has a number of uses, including to access otherwise inaccessible polymer constructions. ‘Given the current monomer pools we have, and mechanisms that we know, we can’t otherwise build certain polymers that we would want to have,’ says Zhukhovitskiy. His group is developing tools for this purpose. In 2021, he reported the transformation of polyesters into vinyl polymers via the Ireland−Claisen sigmatropic rearrangement. The standard retrosynthetic analysis of a vinyl polymer is to start with a vinyl monomer, Zhukhovitskiy explains, but when the desired monomer is unavailable ‘we showed you can make it by the editing process’.
The Zhukhovitskiy group is also looking at skeletal editing to convert robust polymers with only carbon in their backbones into degradable polymers that are easier to recycle. ‘It would be great if we could take a polymer like polyethylene and do an insertion of heteroatoms into its backbone to make it more readily degradable,’ he says.
Not just small molecules
Skeletal editing reactions may have been first dreamt-up to make it easier for medicinal chemists to synthesize analogues of potential drug molecules more quickly, but their utility is starting to be explored in natural product synthesis too. Polymer chemists have also explored skeletal editing in recent years (see Editing polymer backbones box above).
In natural product synthesis, skeletal editing can enable novel reaction pathways that lead to alternative, shorter synthetic routes. In 2021, for example, Mingji Dai, an organic chemist at Emory University in Atlanta, US, reported the use of a Ciamician–Dennstedt rearrangement to shorten the total synthesis of the pyridine-containing alkaloids lycodine and complanadine A. The latter has been identified as a lead compound for developing treatment for neurological disorders and persistent pain management.
Each problem requires different chemical modalities
Dai showed that using a nucleophilic pyrrole ring as a precursor for an electrophilic pyridine allowed chemistries to be used during the early stages of the synthesis that would otherwise be impossible. ‘The pyrrole enabled us to do chemistry which cannot be done on pyridine,’ says Dai.
Using this approach, his team synthesised lycodine in nine steps from a commercially available molecule, compared to the 11–15 steps previously reported, and complanadine A in 12 steps, compared to the 15–20 steps previously reported. For both alkaloids, the Ciamician–Dennstedt rearrangement to create 3-chloropyridine was step seven and the key reactions that came before it were a one-pot Staudinger reduction, an amine–ketone condensation, and Mannich-type cyclization.
As chemists continue building the skeletal editing toolbox, most are hoping their work will lead to a seismic shift in the way organic molecules are synthesised in the future. Levin is keen to encourage others to join in and develop tools for desired reactions that cannot be carried out at present. Adding, deleting and switching single atoms are ‘all very different problems which require different chemistry’, he says. ‘It is not a field that is always going to be solved with carbenes or palladium or photochemistry or whatever. Each problem requires different chemical modalities – this makes it a lot of fun because my lab works on lots of different chemistry.’
Nina Notman is a science writer based in Salisbury, UK







3 readers' comments