Scientists hope their strategy will invigorate cinchona alkaloid-mimic catalysis
Researchers in Japan have developed a practical method for synthesising a form of quinine that does not occur in nature.1 This enantiomer and its derivatives may have applications as organocatalysts.
Quinine is an historically important natural product. In the 1600s, Spanish missionaries observed indigenous people in South America using bark from the cinchona tree to treat malaria then introduced it to Europe. Since its isolation in the early 1800s, the compound has been of considerable scientific interest and its synthesis has an extensive history. Robert Burns Woodward and William von Eggers Doering reported the first formal synthesis of quinine in 19442 and in 2001, Gilbert Stork reported the first stereoselective total synthesis of the enantiomer that occurs in nature, (–)-quinine.3
Since 2001, various research groups have devised asymmetric syntheses of quinine, most recently Nuno Maulide from the University of Vienna in Austria and his colleagues, using a C–H activation strategy.4 Maulide describes quinine as ‘a fascinating natural product’ that is currently experiencing a revival as a target for total synthesis. This is because quinine contains a particularly basic nitrogen atom that can catalyse a broad range of enantioselective reactions.
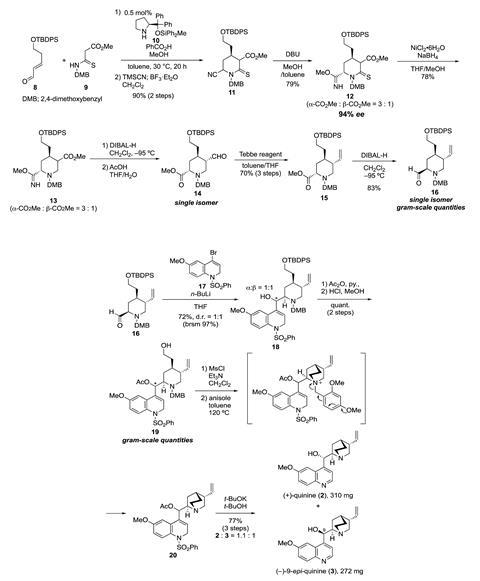
Now, a team of scientists led by Hayato Ishikawa of Kumamoto University has synthesised (+)-quinine, the enantiomer that does not occur in nature, using a practical asymmetric method that gives the target compound in 16% overall yield over 15 steps. Maulide describes the approach as ‘going back to the Hoffmann–La Roche disconnection, where you use a quinoline nucleophile to attack an aldehyde, so that is actually quite interesting – it’s a new take on an old disconnection’.
Ishikawa says using an organocatalytic formal aza [3+3] cycloaddition/Strecker-type cyanation reaction to form a piperidine derivative, with only 0.5 mol% of the organocatalyst, is a particularly significant part of the synthesis. When synthesising the piperidine intermediate, the researchers say ‘it was very difficult to reduce the cyano, thiocarbonyl and ester groups on the piperidine ring in a stepwise fashion’, and they overcame this by forming an imidate group from the cyano functionality.
The next stage was to introduce a quinoline derivative. ‘The researchers cleverly employ a lower oxidation state coupling partner, a dihydroquinoline, to avoid homodimerisation byproducts while improving reactivity in the key fragment coupling reaction. This tactic neatly solves two problems and tickles the brain when considering strategies to access related target molecules,’ comments Jennifer Stockdill who develops new ways to make complex natural products at Wayne State University in the US. The final steps of the synthesis involved a tandem cyclisation–deprotection cascade, that Stockdill calls ‘a beautiful solution to the problem of avoiding alkylation of either a secondary or tertiary amine precursor to the natural product, unveiling the highly basic lone pair at the last possible moment.’
The Ishikawa group are excited to explore the applications of (+)-quinine, and are particularly interested in using it to construct axial and helical chirality. They are looking to improve the synthesis of the compound, describing the ‘five-step total synthesis of (+)-quinine in 50% overall yield’ as their next dream.
References
1 S Shiomi et al, Chem. Sci., 2019, 10, 9433 (DOI: 10.1039/c9sc03879e) (This article is open access.)
2 R B Woodward and W E Doering, J. Am. Chem. Soc., 1944, 66, 5, 849 (DOI: 10.1021/ja01233a516)
3 G Stork et al, J. Am. Chem. Soc., 2001, 123, 14, 3239 (DOI: 10.1021/ja004325r)
4 D H O’Donovan et al, Angew. Chem. Int. Ed., 2018, 57, 10737 (DOI: 10.1002/anie.201804551)








No comments yet