
Directed evolution has created an enzyme that catalyses a challenging carbon–carbon bond formation reaction for synthesising structurally complex antibiotics.
Identified around 20 years ago, arylomycins demonstrate broad-spectrum antibacterial activity but because they are so tricky to make, they are not yet used in the clinic. Composed of a lipid tail and a macrocyclic core, they target a different bacterial process from existing treatments – a valuable addition to our limited antibiotic arsenal.
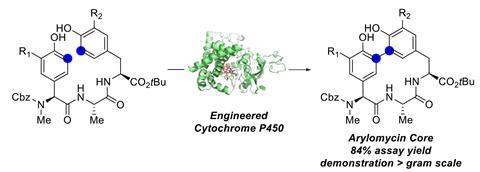
Arylomycins’ macrocycle comprises three amino acids and two phenyl groups, linked by a biaryl bond. Forming this carbon–carbon bond has proven troublesome: traditional methods rely on metal-catalysed cross couplings, which require first installing functional groups to facilitate the reaction and often produce complex product mixtures. Scaling this type of chemistry is challenging, and there are concerns surrounding using potentially toxic metals. However, a team surrounding Carmela Molinaro from Genentech in the US has designed a P450 enzyme to overcome these obstacles.
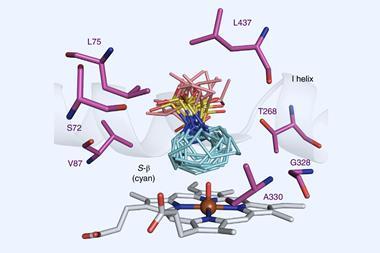
The cytochromes P450 are a superfamily of iron-containing enzymes, found in almost every living organism. These enzymes commonly catalyse hydroxylation reactions, inserting oxygen from the air into carbon–hydrogen bonds to produce alcohols. But researchers have also bioengineered P450 enzymes for other transformations. Such evolved enzymes are incredibly selective, producing single products in high yields, without needing certain functional groups in the substrate.
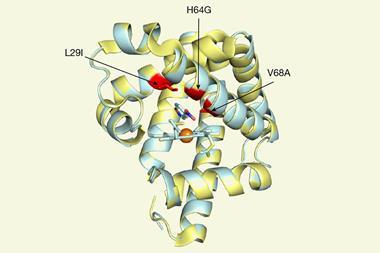
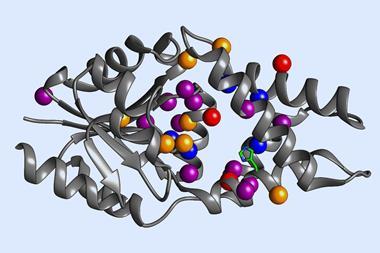
The Genentech researchers’ initial screening identified a number of P450 variants that catalyse a carbon–carbon bond formation on test substrates. Through directed evolution – an iterative process of mutation and screening – the team tailored the enzyme’s reactivity further. Their best performing enzyme catalyses the arylomycin C–C bond formation reaction in 84% yield.
‘It really illustrates the power of enzymatic catalysis to simplify access to important molecules,’ comments Hans Renata, a researcher in chemoenzymatic synthesis from Rice University, US. ‘The previous route used by Genentech to prepare arylomycin relied upon a palladium cross coupling, which requires the use of a pre-functionalised substrate. In contrast, the biocatalytic strategy does not, which translates to easier substrate synthesis.’
The researchers prepared several related arylomycin cores using the same engineered enzymes. Some of these compounds even featured bromide substituents, which would be incompatible with traditional cross coupling reactions.
The Genentech team then investigated scaling up the reaction, a notoriously difficult process in drug development. ‘The demonstration of the reaction on gram scale is very impressive,’ says Renata. ‘The next step should be to show how this transformation can be adapted on process scale.’
References
C Molinaro et al, J. Am. Chem. Soc., 2022, 144, 14838 (DOI: 10.1021/jacs.2c06019)









No comments yet